Версия Seurat V5 считывает данные одной ячейки в разных форматах.
Краткое изложение предыдущей ситуации
В марте 2023 года (я подсознательно думал, что это было в этом году, ххх, но вдруг понял, что это было уже 24 года назад) автор Rookie Group собрал методы чтения одноячеечных данных в разных форматах, которые были основаны на версии V4. .
Читайте данные транскриптома одной клетки в разных форматах и решайте возникающие проблемы.
Когда я изучал отдельные клетки,,Чтение данных основано на методе, указанном в твите.,Вот и всеЗагрузите и прочитайте процесс анализа данных одной ячейки в разных форматах.эта заметка。
ноВ настоящее время пакет seurat обновлен до версии 5.0.1.,После обновления также потребовалось некоторое время для использования.Предварительное исследование по обновлению и использованию пакета Seurat
Хотя это похоже на структуру объекта seurat,V4иV5Большой разницы между версиями нет——Подробное сравнение внутренней структуры объектов Сёра в версиях V5 и V4.,нодаПри чтении данных разница между V4 и V5 все еще немного очевидна.
Если это один образец, просто прочитайте его напрямую и создайте объект seurat.:Первая попытка версии Seurat V5
Основное отличие заключается в,В версии V4 образцы обычно считываются в цикле, CreateSeuratObject используется для создания объекта seurat, а затем для организации данных используется слияние.
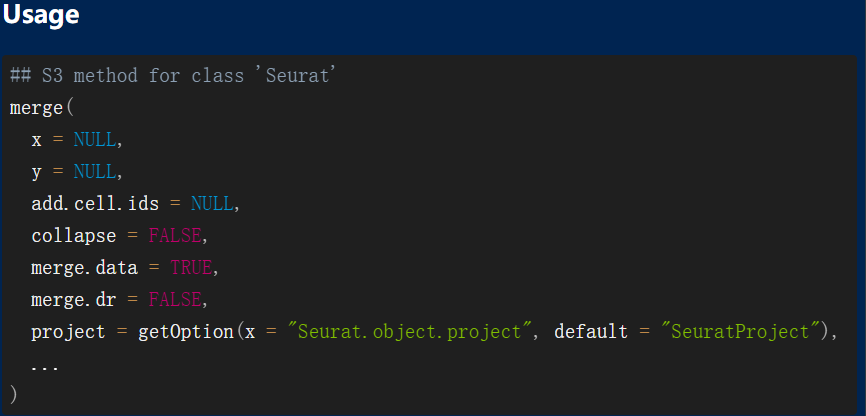
И вВ версии seurat V5, если несколько файлов считываются отдельно, а затем используется функция слияния, матрица выражений каждого образца не объединяется.。тогда мы сможемСначала объедините несколько образцов в сверхбольшую матрицу выражений и назовите строки именами генов, а имена столбцов — информацией штрих-кодов. Затем напрямую используйте функцию CreateSeuratObject для создания объекта Seurat, который идеально подходит для последующего анализа.
Используйте Seurat v5 для чтения нескольких 10-кратных матриц транскриптома одной клетки
Несколько методов чтения данных из одной ячейки в разных форматах
Прежде чем читать данные для анализа, нам необходимо установить и загрузить необходимые пакеты R. В предыдущих твитах также был организован ряд пакетов R, которые необходимо установить.
library(COSG)
library(harmony)
library(ggsci)
library(dplyr)
library(future)
library(Seurat)
library(clustree)
library(cowplot)
library(data.table)
library(dplyr)
library(ggplot2)
library(patchwork)
library(stringr)
стандартный формат 10X
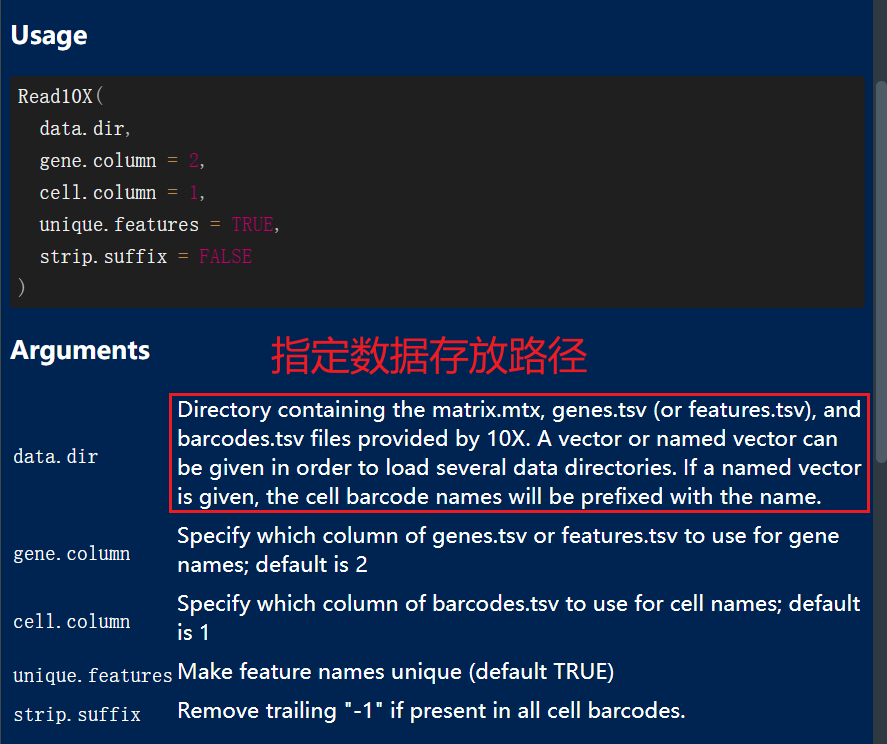
в случаестандартный формат 10Xиз несколькихданные,Затем мы используем функцию Read10X() для чтения нескольких данных.,Затем создайте seuratобъект
##стандартный формат 10X
#Особой разницы в чтении данных V4 и V5 одного образца нет
#липкий
samples=list.files("./GSE212975/")
samples
dir <- file.path('./GSE212975/',samples)
names(dir) <- samples
#readданныеcreateSeuratобъект
counts <- Read10X(data.dir = dir)
sce.all = CreateSeuratObject(counts,
min.cells = 5,
min.features = 300 )
dim(sce.all) #Просмотр количества генов и общего количества клеток
as.data.frame(sce.all@assays$RNA$counts[1:10, 1:2])
table(sce.all@meta.data$orig.ident) #Просмотр количества клеток в каждом образце
head(sce.all@meta.data)
формат h5
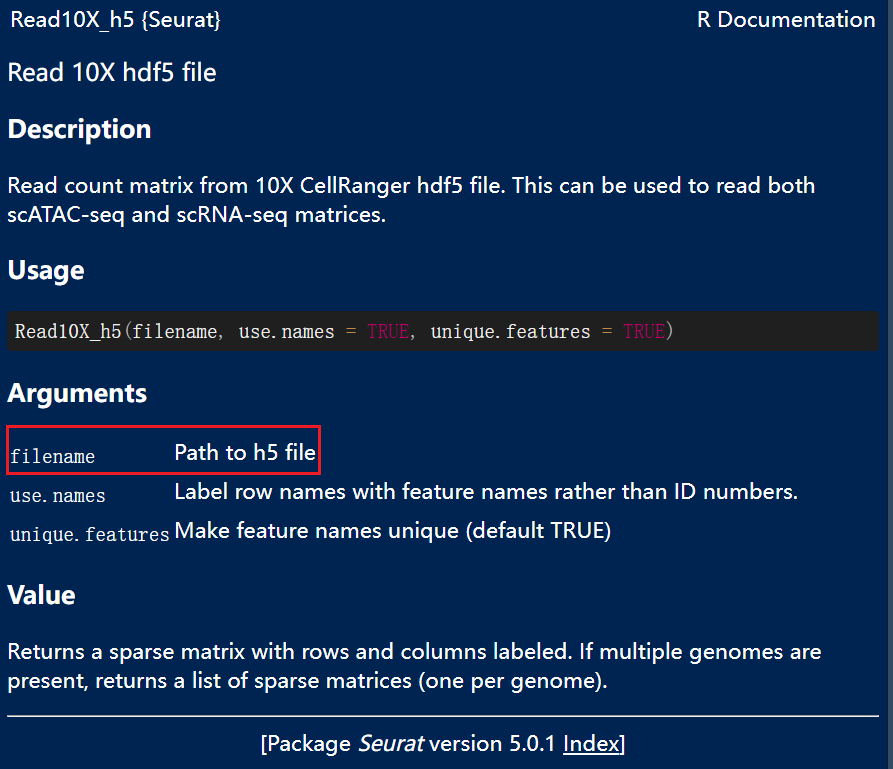
формат h5На самом деле существуют соответствующиеФункция Read10X_h5() может читать напрямую, но Read10X_h5 использует цикл для чтения нескольких файлов данных и возвращает список, который необходимо интегрировать вручную.
#Загружаем необходимые пакеты R
library(hdf5r)
library(stringr)
library(data.table)
#Установить путь к файлу
dir='./GSE159115_RAW/'
samples=list.files( dir )
samples
#читатьформат файл h5
sceList = lapply(samples,function(pro){
print(pro)
counts = Read10X_h5( file.path(dir,pro))
return(counts)
})
#Измените имена столбцов, чтобы сделать штрих-коды конкретными
samples<-str_split(samples,"_",simplify = T)[,1]
for (i in 1:length(sceList)) {
col<-colnames(sceList[[i]])
colnames(sceList[[i]])<-paste0(samples[i],"_",col)
}
#данные Интегрировано для создания seuratобъектов
merge <- do.call(cbind,sceList)
sce =CreateSeuratObject(counts = merge ,
min.cells = 5,
min.features = 300 )
#Просмотр структуры объекта
sce
as.data.frame(sce@assays$RNA$counts[1:10, 1:2])
head(sce@meta.data, 10)
table(sce@meta.data$orig.ident)
форматы txt.gz и csv.gz
Методы чтения форматов txt.gz и csv.gz мало чем отличаются, поэтому они отнесены к одной категории.
Справочный твит:Использование Seurat v5 для чтения нескольких одноячеечных проектов, которые не являются стандартными файлами 10x.
формат txt.gz
dir='./GSE167297/'
samples=list.files( dir ,pattern = 'gz')
samples
library(data.table)
#Сначала прочтите матрицу
ctList = lapply(samples,function(pro){
# pro=samples[1]
print(pro)
ct=fread(file.path( dir ,pro),data.table = F)
ct[1:4,1:4]
rownames(ct)=ct[,1]
colnames(ct) = paste(gsub('_CountMatrix.txt.gz','',pro),
colnames(ct) ,sep = '_')
ct=ct[,-1]
return(ct)
})
#Интегрируем данные в один большой список
lapply(ctList, dim)
tmp =table(unlist(lapply(ctList, rownames)))
cg = names(tmp)[tmp==length(samples)]
bigct = do.call(cbind,
lapply(ctList,function(ct){
ct = ct[cg,]
return(ct)
}))
dim(bigct)
#создать объект безопасности
sce.all=CreateSeuratObject(counts = bigct,
min.cells = 5,
min.features = 300)
sce.all
as.data.frame(sce.all@assays$RNA$counts[1:10, 1:2])
head(sce.all@meta.data, 10)
table(sce.all@meta.data$orig.ident)
формат csv.gz
library(data.table)
dir='./GSE129516_RAW/'
samples=list.files( dir )
samples
sceList = lapply(samples,function(pro){
# pro=samples[1]
print(pro)
ct=fread( file.path(dir,pro),data.table = F)
ct[1:4,1:4]
ct[nrow(ct),1:4]
rownames(ct)=ct[,1]
colnames(ct) = paste(gsub('_filtered_gene_bc_matrices.csv.gz','',pro),
colnames(ct) ,sep = '_')
ct=ct[,-1]
ct[1:4,1:4]
return(ct)
})
lapply(sceList, dim)
tmp =table(unlist(lapply(sceList, rownames)))
cg = names(tmp)[tmp==length(samples)]
bigct = do.call(cbind,
lapply(sceList,function(ct){
ct = ct[cg,]
return(ct)
}))
dim(bigct)
#создать объект безопасности
sce.all=CreateSeuratObject(counts = sceList,
min.cells = 5,
min.features = 300)
sce.all
as.data.frame(sce.all@assays$RNA$counts[1:10, 1:2])
head(sce.all@meta.data, 10)
table(sce.all@meta.data$orig.ident)
Подводя итог, вот что мы узнали на данный момент: методов чтения данных из одной ячейки в разных форматах Ла!
Небольшой особый случай 10X
Вообще говоря, стандартному формату 10X по названию образца будут соответствовать три файла: barcodes.tsv.gz, Features.tsv.gz, matrix.mtx.gz.,ноGSE184708За некоторыми незначительными исключениями,Хотя образцов двадцать,Но данные интегрированы
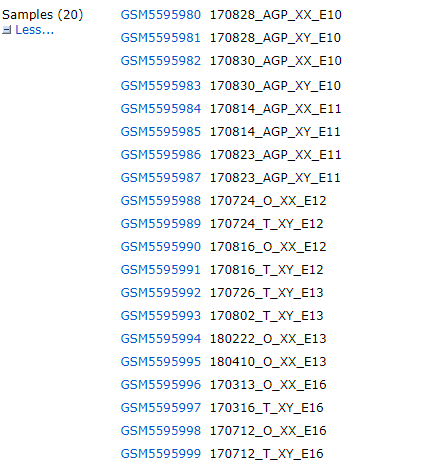
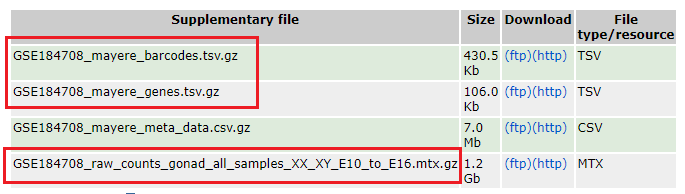
После загрузки данных считайте штрих-коды, гены и файлы матричной матрицы соответственно, организуйте три файла в стандартизированную матрицу с именами строк и столбцов, а затем создайте объект seurat.
#Загружаем необходимые пакеты R
library(data.table)
library(Matrix)
#Прочитайте три файла в соответствии с соответствующими форматами.
mtx=readMM( "./GSE184708/GSE184708_raw_counts_gonad_all_samples_XX_XY_E10_to_E16.mtx.gz" )
mtx[1:4,1:4]
dim(mtx)
cl=fread( "./GSE184708/GSE184708_mayere_barcodes.tsv.gz" ,
header = F,data.table = F )
head(cl)
rl=fread( "./GSE184708/GSE184708_mayere_genes.tsv.gz" ,
header = F,data.table = F )
head(rl)
#Интегрировать информацию матрицы
colnames(mtx)=cl$V1
rownames(mtx)=rl$V1
#создать объект безопасности
sce.all=CreateSeuratObject(counts = mtx ,
project = 'mouse',
min.cells = 5,
min.features = 300 )
#Группа
as.data.frame(sce.all@assays$RNA$counts[1:10, 1:2])
head(sce.all@meta.data, 10)
table(sce.all$orig.ident)
library(stringr)
phe=str_split(rownames(sce.all@meta.data),'_',simplify = T)
head(phe)
table(phe[,2])
sce.all$group=phe[,2]
table(phe[,3])
sce.all$sex=phe[,3]
table(phe[,1])



Неразрушающее увеличение изображений одним щелчком мыши, чтобы сделать их более четкими артефактами искусственного интеллекта, включая руководства по установке и использованию.
Копикодер: этот инструмент отлично работает с Cursor, Bolt и V0! Предоставьте более качественные подсказки для разработки интерфейса (создание навигационного веб-сайта с использованием искусственного интеллекта).
Новый бесплатный RooCline превосходит Cline v3.1? ! Быстрее, умнее и лучше вилка Cline! (Независимое программирование AI, порог 0)

Разработав более 10 проектов с помощью Cursor, я собрал 10 примеров и 60 подсказок.
Я потратил 72 часа на изучение курсорных агентов, и вот неоспоримые факты, которыми я должен поделиться!

Идеальная интеграция Cursor и DeepSeek API
DeepSeek V3 снижает затраты на обучение больших моделей
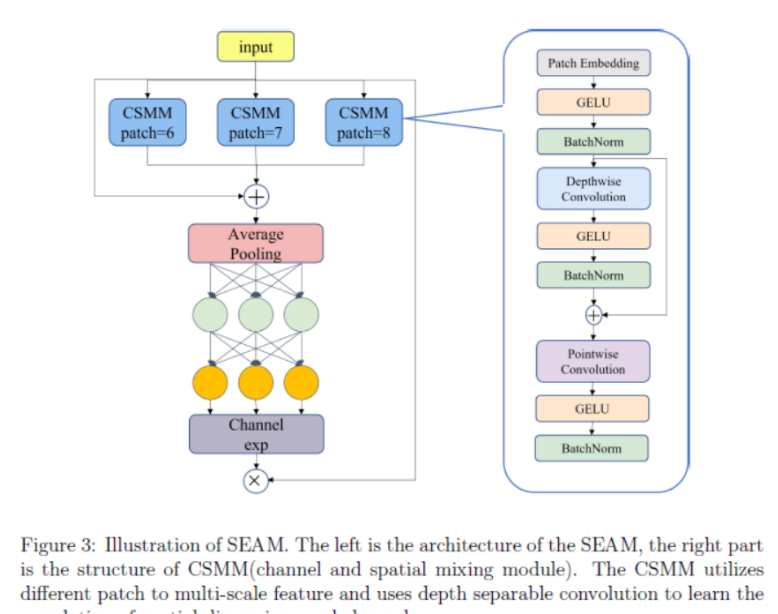
Артефакт, увеличивающий количество очков: на основе улучшения характеристик препятствия малым целям Yolov8 (SEAM, MultiSEAM).

DeepSeek V3 раскручивался уже три дня. Сегодня я попробовал самопровозглашенную модель «ChatGPT».

Open Devin — инженер-программист искусственного интеллекта с открытым исходным кодом, который меньше программирует и больше создает.
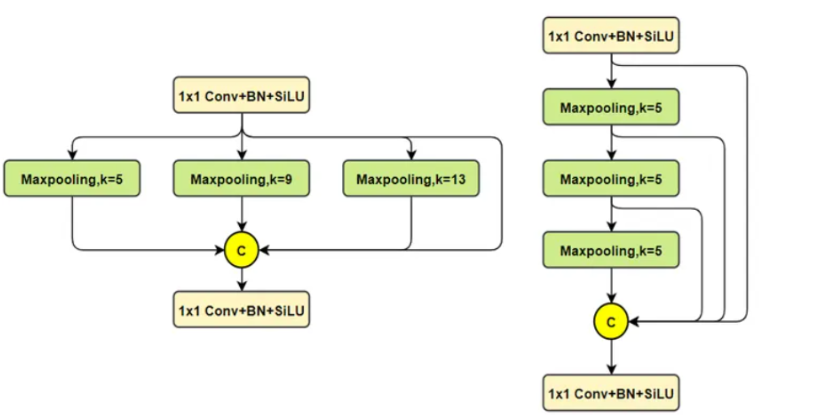
Эксклюзивное оригинальное улучшение YOLOv8: собственная разработка SPPF | SPPF сочетается с воспринимаемой большой сверткой ядра UniRepLK, а свертка с большим ядром + без расширения улучшает восприимчивое поле

Популярное и подробное объяснение DeepSeek-V3: от его появления до преимуществ и сравнения с GPT-4o.

9 основных словесных инструкций по доработке академических работ с помощью ChatGPT, эффективных и практичных, которые стоит собрать
Вызовите deepseek в vscode для реализации программирования с помощью искусственного интеллекта.
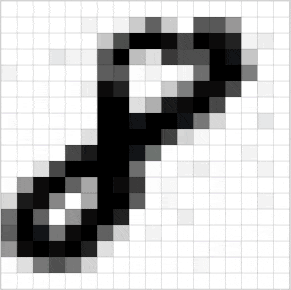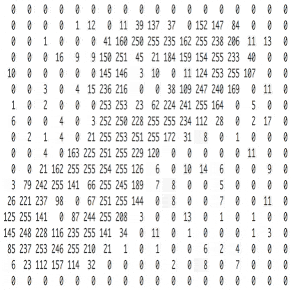
Познакомьтесь с принципами сверточных нейронных сетей (CNN) в одной статье (суперподробно)
50,3 тыс. звезд! Immich: автономное решение для резервного копирования фотографий и видео, которое экономит деньги и избавляет от беспокойства.
Cloud Native|Практика: установка Dashbaord для K8s, графика неплохая
Краткий обзор статьи — использование синтетических данных при обучении больших моделей и оптимизации производительности
MiniPerplx: новая поисковая система искусственного интеллекта с открытым исходным кодом, спонсируемая xAI и Vercel.
Конструкция сервиса Synology Drive сочетает проникновение в интрасеть и синхронизацию папок заметок Obsidian в облаке.
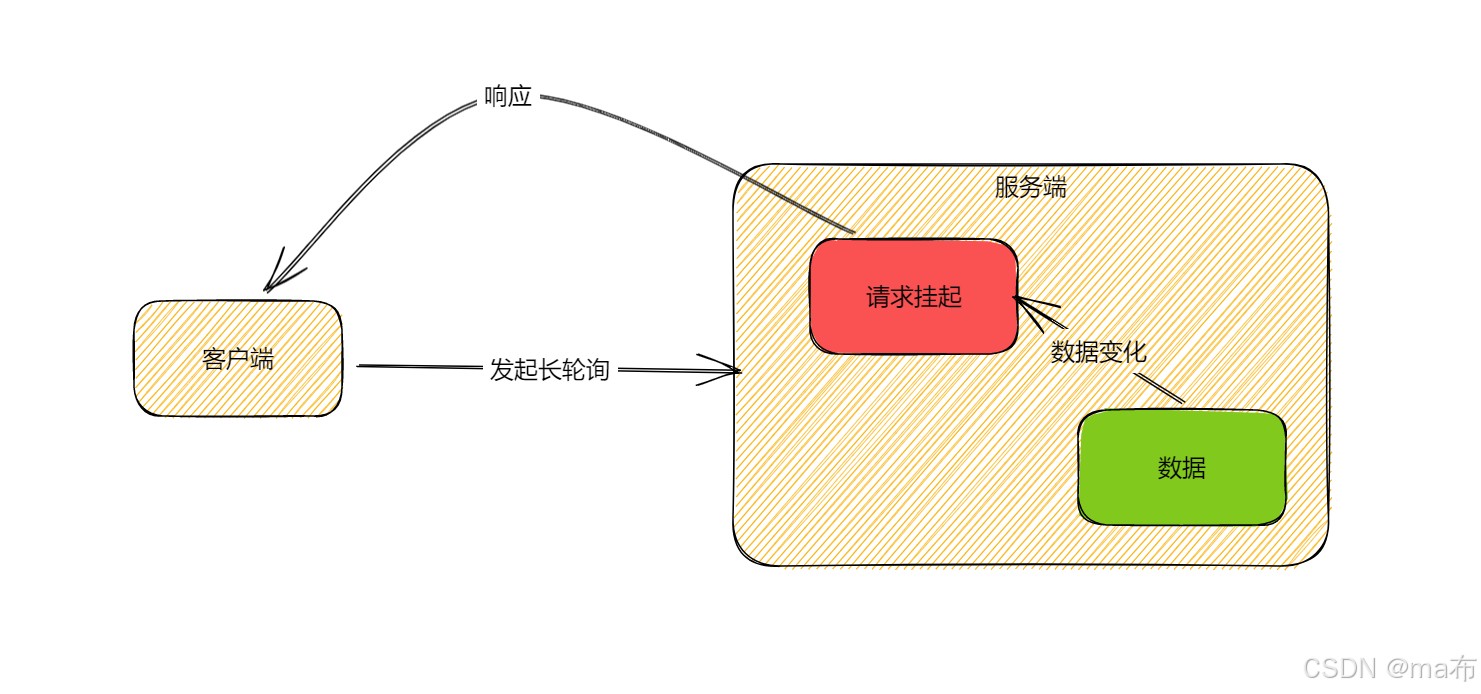
Центр конфигурации————Накос
Начинаем с нуля при разработке в облаке Copilot: начать разработку с минимальным использованием кода стало проще

[Серия Docker] Docker создает мультиплатформенные образы: практика архитектуры Arm64

Обновление новых возможностей coze | Я использовал coze для создания апплета помощника по исправлению домашних заданий по математике

Советы по развертыванию Nginx: практическое создание статических веб-сайтов на облачных серверах
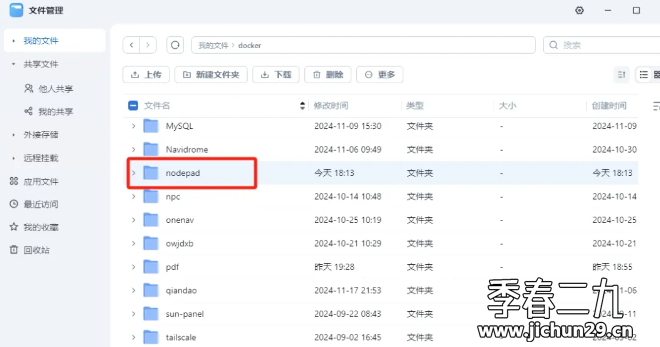
Feiniu fnos использует Docker для развертывания личного блокнота Notepad

Сверточная нейронная сеть VGG реализует классификацию изображений Cifar10 — практический опыт Pytorch
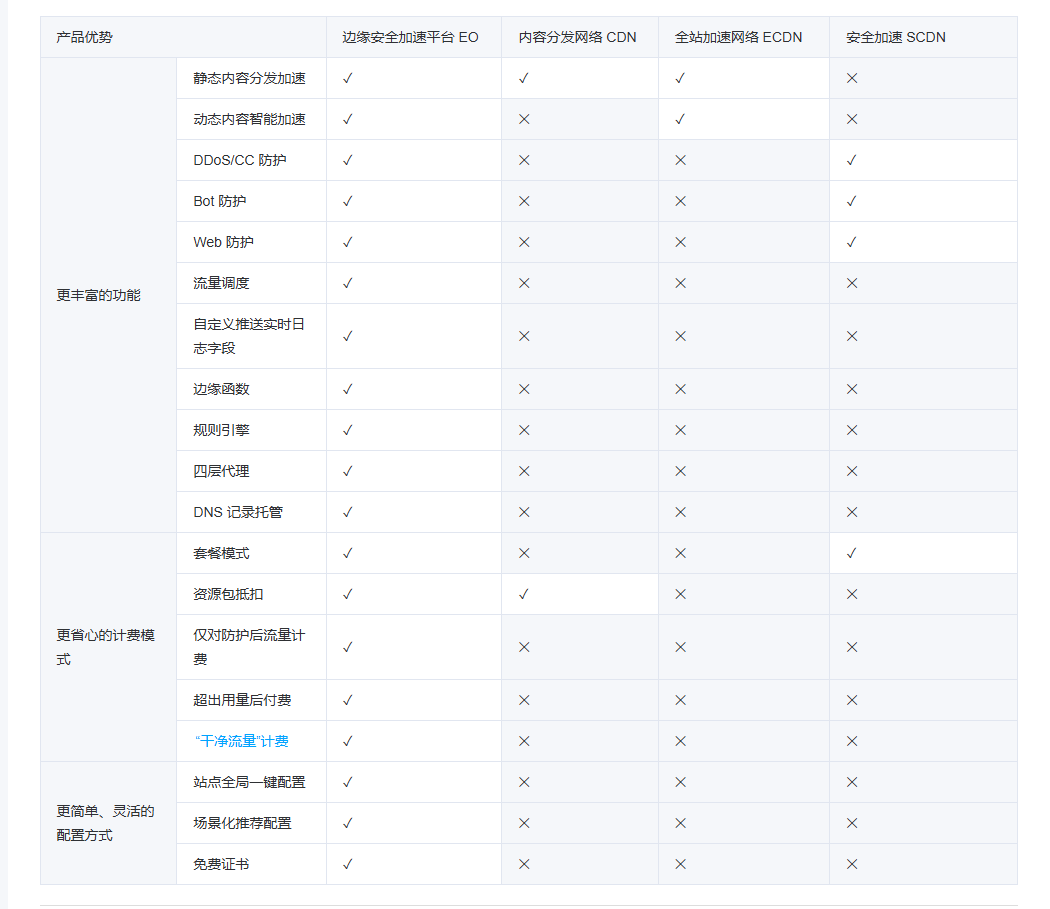
Начало работы с EdgeonePages — новым недорогим решением для хостинга веб-сайтов
[Зона легкого облачного игрового сервера] Управление игровыми архивами