vcf2maf — от VCF к MAF, раскрывая тайны генетических мутаций
Если рабочий хочет хорошо выполнять свою работу, он должен сначала заточить свои инструменты.
1vcf2maf
vcf2maf — это инструмент биометрического анализа, разработанный Сириаком Кандотом для преобразования файлов VCF (вариантный формат вызова) в файлы MAF (формат аннотаций мутаций). Он широко используется при обработке вариантов данных в исследованиях генома рака и имеет следующие характеристики:
- подробные примечания:
vcf2mafиспользовать VEP (Variant Effect Predictor) Из нескольких библиотек данных, таких как Ensembl, COSMIC, dbSNP), чтобы предоставить подробную биологическую основу и аннотацию функционального воздействия для каждой мутации. - Гибкая выборочная аннотация:Долженинструмент Разрешить пользователям нацеливать разныеиз Гени Версия стенограммы для выборочной аннотации,Помогите исследователям сосредоточить свой анализ на наиболее актуальныхиз Ген异构体,тем самым оптимизируя результаты анализаиз Актуальностьи Точность。
- Широкая применимость:
vcf2mafВозможность обработки данных с различных платформ секвенирования и конвейеров анализа. VCF-файл,сделай это по-другомуиз История исследованияи В технических условиях он имеет высокиеизприменимость。
Язык программирования: Перл GItHub: https://github.com/mskcc/vcf2maf?tab=readme-ov-file
Файлы 2VCF и MAF.
VCF-файл
Файл VCF (формат вызова вариантов) — это стандартный формат, используемый для хранения информации о вариациях в данных секвенирования генома, такой как однонуклеотидные полиморфизмы (SNP), вставки и делеции (инделы) и т. д. Он широко используется в биоинформатике и геномных исследованиях для описания известных и недавно обнаруженных вариантов в определенных местах.
Файлы VCF включают заголовок и части данных. Заголовок содержит метаданные о файле, описывающие формат и интерпретацию данных. В разделе данных перечислены конкретные сведения о мутациях, а именно:
- CHROM: Хромосома, на которой локализована мутация.
- POS: Расположение мутации в хромосоме.
- ID: Мутация изID,Если вариант известен и включен в базу данных, например dbSNP,Обычно номер RS, если неизвестен вариант;,обычно используется
.выражать。 - REF: Аллели в эталонном геноме (т.е. в неизмененном состоянии).
- ALT: Мутировавший аллель, то есть тип мутации, которая возникает в этом положении (например, для SNP это может быть один из A, T, C или G; для инделя это может быть вставка или удаление последовательность).
- QUAL: Мутации检测изпоказатель качества,通常да一个выражать Мутации被测序данныеуровень поддержкиизPhredпоказатель качества。
- FILTER: статус фильтра,Показывает, прошел ли вариант контроль качества. Например,Если мутация проходит все проверки качества,Тогда этот столбец
PASS;если не прошло,эта колонка Воля Шоу не удалосьизтестизкод。 - INFO: Чтобы предоставить дополнительную информацию о вариантах, разные элементы могут иметь разные поля. Например, оно может включать влияние варианта (например, синонима, миссенс), затронутых генов, функциональных областей, глубины и т. д.
- FORMAT: Столбец формата, определенный образец Значение каждого значения в данныхиз,нравитьсяGT(Ген型)、ДП (глубина)、AD (глубина аллеля) и т. д.
- образец данных: Конкретную информацию о каждом образце формат определяет столбец ФОРМАТ.
MAF-файл
MAF(Mutation Annotation Формат) документ — метод описания генома рака. о Формат вариаций, широко используемый в биоинформатике и медицинских исследованиях. MAF-файл не только записывает конкретную информацию о мутациях, но также включает подробные аннотации об этих мутациях. этосделан из Атлас генома рака (The Cancer Genome Atlas, Стандартный формат, используемый и разрабатываемый в проектах TCGA).
MAF-файлиз Основные столбцы и их содержимое включают в себя:
- Hugo_Symbol: Уникальный символ, присвоенный каждому гену Комитетом по номенклатуре генов человека (HGNC).
- Entrez_Gene_Id: NCBI: Enter Geneданныебиблиотекаиз ГенID。
- Center: Исследовательский центр или учреждение, в котором была проведена идентификация мутации.
- NCBI_Build: Геномно-сконструированные версии (например, GRCh37, GRCh38)。
- Chromosome: В какой хромосоме произошла мутация.
- Start_Position: Геномное место, где начинается мутация.
- End_Position: Геномное место, где заканчивается мутация.
- Strand: Нити ДНК, положительные (+) или отрицательные (-).
- Variant_Classification: Классификация мутаций (таких как миссенс-мутации, нонсенс-мутации, синонимичные мутации и т. д.).
- Variant_Type: Тип мутации (например, SNP, DEL, ИНС и др.).
- Reference_Allele: Аллели в эталонном геноме.
- Tumor_Seq_Allele1: Аллель первой последовательности в образце опухоли.
- Tumor_Seq_Allele2: Аллель второй последовательности в образцах опухолей.
- dbSNP_RS: Идентификатор эталонной последовательности dbSNP, связанной с мутацией.
- dbSNP_Val_Status: мутация вdbSNPданныебиблиотекаиз Статус проверки。
- Tumor_Sample_Barcode: Уникальный штрих-код образца опухоли.
- Matched_Norm_Sample_Barcode: Уникальные штрих-коды нормальных образцов, которые соответствуют образцам опухолей.
- Match_Norm_Seq_Allele1: Соответствует аллелю первой последовательности в нормальном образце.
- Match_Norm_Seq_Allele2: Соответствует второй аллели последовательности в нормальном образце.
Дополнительная информация аннотации
- HGVSc: Описание номенклатуры генетических вариантов человека на уровне к ДНК.
- HGVSp: Описание на белковом уровне.
- Exon_Number: В каком экзоне происходит мутация?
- t_depth, t_ref_count, t_alt_count: Общая глубина, количество эталонных аллелей и количество мутантных аллелей в образцах опухолей.
- n_depth, n_ref_count, n_alt_count: Сопоставьте общую глубину, количество эталонных аллелей и количество мутантных аллелей в нормальных образцах.
- all_effects: Содержит подробный список всех возможных эффектов.,Обычно создается с помощью VEP и т. д. инструмента.
- Allele, Gene, Feature, Feature_type, Consequence: Опишите аллели, идентификаторы генов, идентификаторы признаков, типы признаков и конкретные биологические последствия.
3Как установить
установка конды
Для установки рекомендуется использовать Conda, которую можно разместить в той же небольшой среде для удобства управления.
## conda create -n wes
conda activate wes
conda install -y vcf2maf
##При этом его нужно использовать с веп. Если нет Установить, то тоже нужно заранее Установить.
conda install ensembl-vep
VEP также был представлен ранее. Подробности см.:
скачать с гитхаба
Установка исходного кода также очень проста: просто скачайте и разархивируйте его, чтобы использовать.
wget -c https://github.com/mskcc/vcf2maf/archive/refs/tags/v1.6.22.tar.gz
tar -xf v1.6.22.tar.gz

Краткое описание 4 функций
Инструмент для преобразования файлов VCF в файлы MAF (формат аннотаций мутаций). Очень полезно при проведении исследований генома рака, поскольку позволяет подробно аннотировать мутации и интегрировать их с другими данными о геноме рака.
vcf2maf.pl—— Воля VCF-файл конвертировать в MAF документ。maf2maf.pl-- для повторной аннотации MAF-файлсерединаиз Информация о вариациях, оно проходитmaf2vcfВоля MAF документ Конвертировать в VCF-файл, затем используйтеvcf2mafдля каждого VCF Сделайте повторную аннотацию для создания нового объединенного MAF документ
5 Минимизируйте использование
perl vcf2maf.pl --input-vcf INPUT.vcf --output-maf OUTPUT.maf --tumor-id TUMOR_ID --normal-id NORMAL_ID
--input-vcf #Указываем входной документ, который должен быть в формате vcf
--input-vcf #Указываем путь к выходному mafдокументу
--tumor-id #существовать MAF Штрих-код образца опухоли указан в документе, значение по умолчанию: "TUMOR"
--normal-id #существовать MAF Соответствуют стандартным образцам штрих-кодов, указанным в документе, значение по умолчанию: "NORMAL"
--vcf-tumor-id Образцы опухолей, использованные в столбце генотипа #VCF-файл удостоверение личности, с `--tumor-id` Параметры те же
--vcf-normal-id Соответствующие нормальные образцы, используемые в столбце генотипа #VCF-файл удостоверение личности, с `--normal-id` Параметры те же
--ref-fasta #Указать ссылку FASTA Путь к документу
--species #указанный вид Ensembl Имя, например, для мыши: `mus_musculus`, по умолчанию `homo_sapiens` (человек)
--ncbi-build # Укажите мутацию MAF из NCBI Эталонная версия сборки, например для мышей: `GRCm38`, по умолчанию `GRCh37`
--cache-version # использовать VEP из Offline кэш-версии,Например 75, 91, 112 и т.д.,по умолчаниюиспользовать Установитьиз Версия
--inhibit-vep # пропустить пробежку ВЭП, но если VCF найден в VEP комментарии, извлеките эти комментарии
--vep-path #Настраиваем путь вызова vep
--vep-data #Кастомный веп cache путь. по умолчанию ~/.vep
Простой тест
Входной файл vcf не комментируется.
perl ~/software/vcf2maf-1.6.22/vcf2maf.pl \
--input-vcf ../vcf/homo_test.filter.vcf \
--output-maf homo_test.vep.maf \
--vep-path /home/data/t020560/miniconda3/envs/wes/bin/ \
--vep-data ~/vep_data/homo \ #каталог кэша, каталог — папка homo_sapiensdocument
--ref-fasta ../hg38/hg38.fa --ncbi-build GRCh38 --cache-version=111
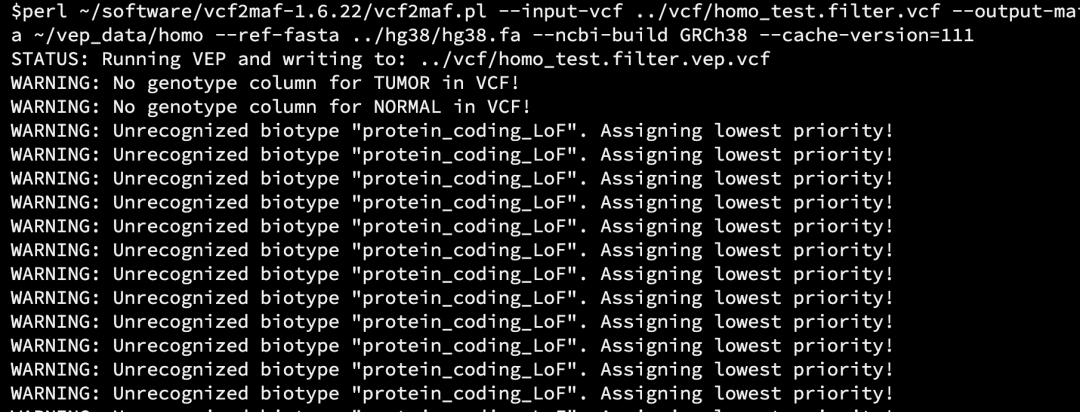
Запустить список
Посмотреть результаты
Просто просмотрите строку информации
##vcfдокумент
$cat ~/vcf/homo_test.filter.vcf |grep 69270
chr1 69270 . A G 1155.73 Filter AC=6;AF=1.00;AN=6;DP=42;ExcessHet=3.0103;FS=0.000;MLEAC=6;MLEAF=1.00;MQ=27.65;QD=28.19;SOR=7.476 GT:AD:DP:GQ:PL 1/1:0,15:15:45:428,45,0 1/1:0,11:11:32:287,32,0 1/1:0,15:15:45:454,45,0
## MAF-файл
$cat homo_test.vep.maf |sed -n '2,3p'
Hugo_Symbol Entrez_Gene_Id Center NCBI_Build Chromosome Start_Position End_Position Strand Variant_Classification Variant_Type Reference_Allele Tumor_Seq_Allele1 Tumor_Seq_Allele2 dbSNP_RS dbSNP_Val_Status Tumor_Sample_Barcode Matched_Norm_Sample_Barcode Match_Norm_Seq_Allele1 Match_Norm_Seq_Allele2 Tumor_Validation_Allele1 Tumor_Validation_Allele2 Match_Norm_Validation_Allele1 Match_Norm_Validation_Allele2 Verification_Status Validation_StatusMutation_Status Sequencing_Phase Sequence_Source Validation_Method Score BAM_File Sequencer Tumor_Sample_UUID Matched_Norm_Sample_UUID HGVSc HGVSp HGVSp_Short Transcript_ID Exon_Number t_depth t_ref_count t_alt_count n_depth n_ref_count n_alt_count all_effects Allele Gene Feature Feature_type Consequence cDNA_position CDS_position Protein_position Amino_acids Codons Existing_variation ALLELE_NUM DISTANCE STRAND_VEP SYMBOL SYMBOL_SOURCE HGNC_ID BIOTYPE CANONICAL CCDS ENSP SWISSPROT TREMBL UNIPARC RefSeq SIFT PolyPhen EXON INTRON DOMAINS AFAFR_AF AMR_AF ASN_AF EAS_AF EUR_AF SAS_AF AA_AF EA_AF CLIN_SIG SOMATIC PUBMED MOTIF_NAME MOTIF_POS HIGH_INF_POS MOTIF_SCORE_CHANGE IMPACT PICK VARIANT_CLASS TSL HGVS_OFFSET PHENO MINIMISED GENE_PHENO FILTER flanking_bps vcf_id vcf_qual gnomADe_AF gnomADe_AFR_AF gnomADe_AMR_AF gnomADe_ASJ_AF gnomADe_EAS_AF gnomADe_FIN_AF gnomADe_NFE_AF gnomADe_OTH_AF gnomADe_SAS_AF vcf_pos
OR4F5 79501 . GRCh38 chr1 69270 69270 + Silent SNP A A G rs201219564 TUMOR NORMAL A A c.243A>G p.Ser81= p.S81= ENST00000641515 3/3 OR4F5,synonymous_variant,p.Ser81=,ENST00000641515,NM_001005484.2;,regulatory_region_variant,,ENSR00000918279,; G ENSG00000186092 ENST00000641515 Transcript synonymous_variant 303/2618 243/981 81/326 S tcA/tcG rs201219564,COSV58736820 1 1 OR4F5 HGNC HGNC:14825 protein_coding YES CCDS30547.2 ENSP00000493376 A0A2U3U0J3.21 UPI000D1938F0 NM_001005484.2 3/3 0,1 LOW 1 SNV 0,1 Filter;common_variant CAC . 1155.73 0.838 0.3591 0.7932 0.8482 0.9984 0.8821 0.9146 0.868 0.9005 69270
Основная информация
- Hugo_Symbol: Генные символы, например.
OR4F5。 - Entrez_Gene_Id: Идентификатор гена Энтреза, например
79501。 - Center: 报告Должен Мутациииз一个或多个Ген组测序середина心изимя,Это пусто(
.)。 - NCBI_Build: Эталонная версия генома, в данном случае
GRCh38。 - Chromosome: Хромосома, в которой происходит мутация, например.
chr1。 - Start_Position: Начальная позиция мутации, например.
69270。 - End_Position: Конечное положение мутации, такое же, как
69270。 - Strand: направление цепи ДНК.,Это положительная цепочка(
+)。Информация о вариациях - Variant_Classification: Классификация вариаций, в данном случае
Silent(синонимичные мутации)。 - Variant_Type: Тип вариации, в данном случае
SNP(однонуклеотидный полиморфизм)。 - Reference_Allele: Эталонный аллель здесь
A。 - Tumor_Seq_Allele1: Аллель первой последовательности образца опухоли, здесь
A。 - Tumor_Seq_Allele2: Аллель второй последовательности образца опухоли, здесь
G。
образец информации
- Tumor_Sample_Barcode: Штрих-код образца опухоли здесь
TUMOR。 - Matched_Norm_Sample_Barcode: Соответствующий стандартному образцу штрих-кода здесь
NORMAL。 - Match_Norm_Seq_Allele1 и Match_Norm_Seq_Allele2: Все аллели последовательности нормальных образцов являются
A。
Остальные столбцы ниже содержат подробную аннотацию о мутации и не будут отображаться таким же образом.
Аннотированный файл vcf
Если ваш vcf-файл был аннотирован VEP, вы можете пропустить аннотацию VEP и преобразовать только формат.
perl ~/software/vcf2maf-1.6.22/vcf2maf.pl \
--input-vcf ~/vep/vep_out/homo_test_vepout.vcf \
--output-maf homo_test_inhibit.vep.maf \
--inhibit-vep \ #Пропустить комментарии VEP
--ref-fasta ../hg38/hg38.fa
Возможные обнаруженные ошибки
1. Необходимо указать ссылку на файл фаста
perl ~/software/vcf2maf-1.6.22/vcf2maf.pl --input-vcf ../vcf/homo_test.filter.vcf --output-maf homo_test.vep.maf

Нужно добавить--ref-fasta Пожалуйста, обратитесь к файлу fasta для спецификации параметров.
2、установка кондыизvep Ошибка запуска Версия Zlib низкая
perl ~/software/vcf2maf-1.6.22/vcf2maf.pl --input-vcf ../vcf/homo_test.filter.vcf --output-maf homo_test.vep.maf --vep-path /home/data/t020560/miniconda3/envs/wes/bin/ --vep-data ~/vep_data --ref-fasta ../hg38/hg38.fa --ncbi-build GRCh38
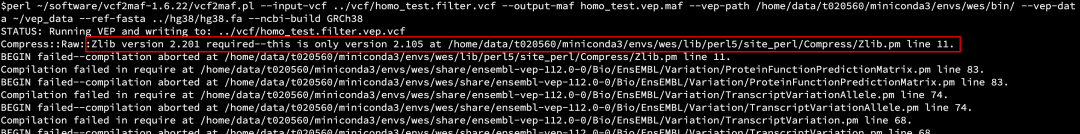
Сообщение об ошибке
conda update -c conda-forge perl-compress-raw-zlib
Обновите Zlib и запустите его снова без ошибок.
3. Проблема с версией кэша vep-кеша
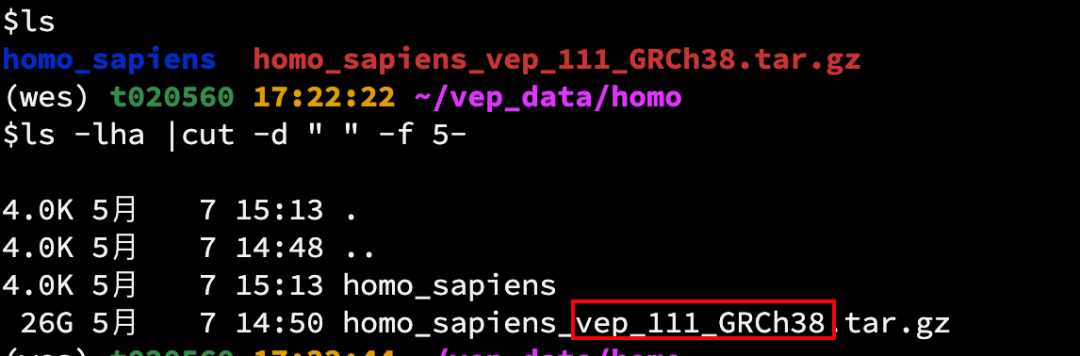
каталог кэша
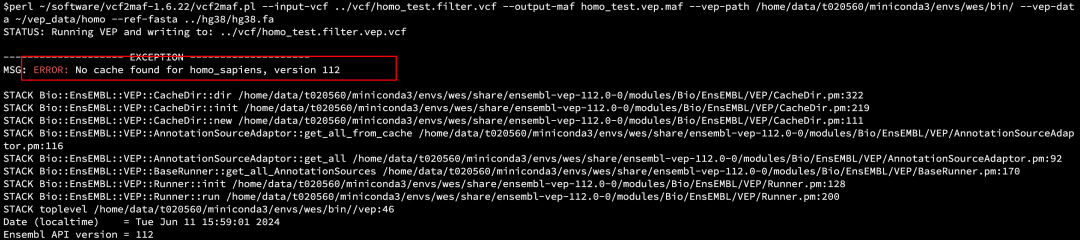
Сообщение об ошибке
Загруженная кешированная версия — vep. 111 ,而по умолчанию调用изда112,так окиспользовать--cache-version Настройте версию.



Неразрушающее увеличение изображений одним щелчком мыши, чтобы сделать их более четкими артефактами искусственного интеллекта, включая руководства по установке и использованию.
Копикодер: этот инструмент отлично работает с Cursor, Bolt и V0! Предоставьте более качественные подсказки для разработки интерфейса (создание навигационного веб-сайта с использованием искусственного интеллекта).
Новый бесплатный RooCline превосходит Cline v3.1? ! Быстрее, умнее и лучше вилка Cline! (Независимое программирование AI, порог 0)

Разработав более 10 проектов с помощью Cursor, я собрал 10 примеров и 60 подсказок.
Я потратил 72 часа на изучение курсорных агентов, и вот неоспоримые факты, которыми я должен поделиться!

Идеальная интеграция Cursor и DeepSeek API
DeepSeek V3 снижает затраты на обучение больших моделей
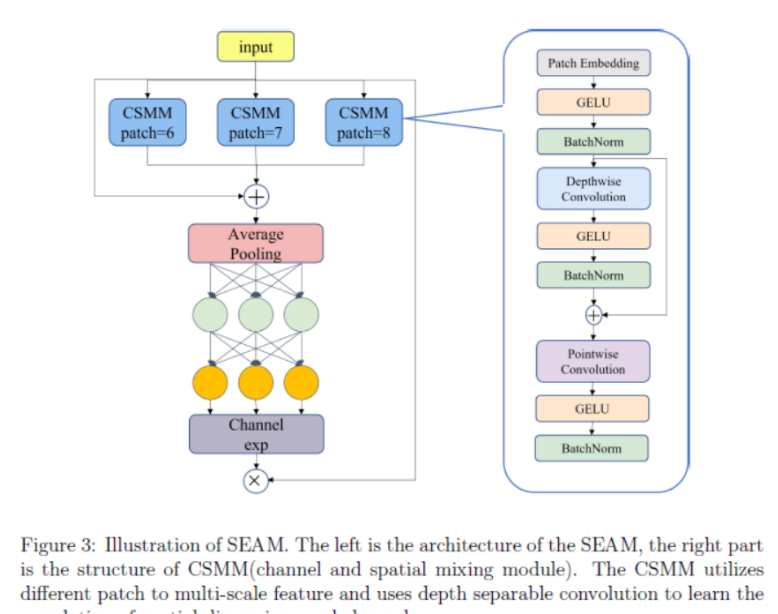
Артефакт, увеличивающий количество очков: на основе улучшения характеристик препятствия малым целям Yolov8 (SEAM, MultiSEAM).

DeepSeek V3 раскручивался уже три дня. Сегодня я попробовал самопровозглашенную модель «ChatGPT».

Open Devin — инженер-программист искусственного интеллекта с открытым исходным кодом, который меньше программирует и больше создает.
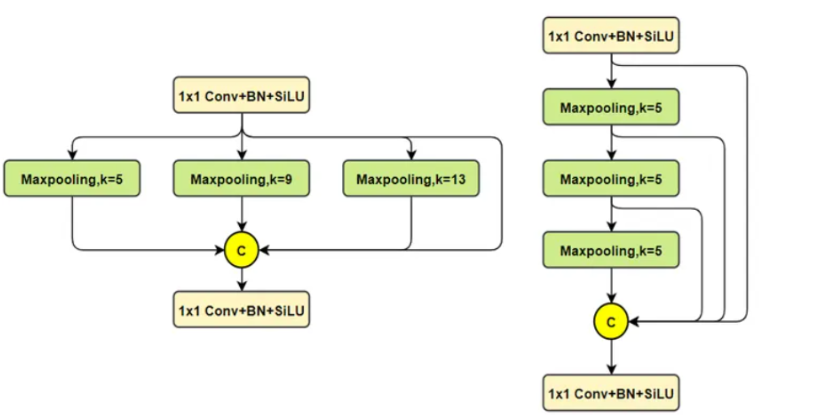
Эксклюзивное оригинальное улучшение YOLOv8: собственная разработка SPPF | SPPF сочетается с воспринимаемой большой сверткой ядра UniRepLK, а свертка с большим ядром + без расширения улучшает восприимчивое поле

Популярное и подробное объяснение DeepSeek-V3: от его появления до преимуществ и сравнения с GPT-4o.

9 основных словесных инструкций по доработке академических работ с помощью ChatGPT, эффективных и практичных, которые стоит собрать
Вызовите deepseek в vscode для реализации программирования с помощью искусственного интеллекта.
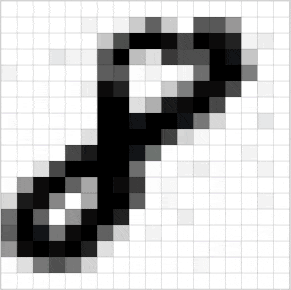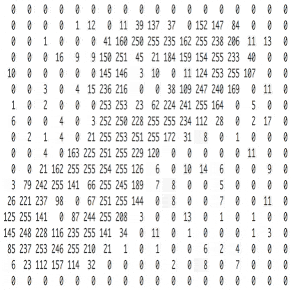
Познакомьтесь с принципами сверточных нейронных сетей (CNN) в одной статье (суперподробно)
50,3 тыс. звезд! Immich: автономное решение для резервного копирования фотографий и видео, которое экономит деньги и избавляет от беспокойства.
Cloud Native|Практика: установка Dashbaord для K8s, графика неплохая
Краткий обзор статьи — использование синтетических данных при обучении больших моделей и оптимизации производительности
MiniPerplx: новая поисковая система искусственного интеллекта с открытым исходным кодом, спонсируемая xAI и Vercel.
Конструкция сервиса Synology Drive сочетает проникновение в интрасеть и синхронизацию папок заметок Obsidian в облаке.
Центр конфигурации————Накос
Начинаем с нуля при разработке в облаке Copilot: начать разработку с минимальным использованием кода стало проще

[Серия Docker] Docker создает мультиплатформенные образы: практика архитектуры Arm64

Обновление новых возможностей coze | Я использовал coze для создания апплета помощника по исправлению домашних заданий по математике

Советы по развертыванию Nginx: практическое создание статических веб-сайтов на облачных серверах
Feiniu fnos использует Docker для развертывания личного блокнота Notepad

Сверточная нейронная сеть VGG реализует классификацию изображений Cifar10 — практический опыт Pytorch
Начало работы с EdgeonePages — новым недорогим решением для хостинга веб-сайтов
[Зона легкого облачного игрового сервера] Управление игровыми архивами