Природа Раскрытие функциональной темной материи посредством глобальной метагеномики
Составлено | Цзэн Цюаньчэнь Рецензент | Ван Цзяньминь
Сегодня я представляю вам статью команды Никоса К. Кирпидеса. Метагеномы содержат огромное количество разнообразных белковых последовательностей, отражающих множество функций и активностей. В прошлом мы обычно исследовали эти пространства последовательностей посредством сравнительного анализа последовательностей в метагеномах с эталонными микробными геномами и семействами белков, полученными из этих геномов. Однако ограничением этого подхода является то, что он может исследовать только известное функциональное разнообразие относительно эталонного генома. Чтобы преодолеть это ограничение и изучить еще больше неизвестного функционального разнообразия, авторы разработали вычислительный метод для создания семейств белков из пространства последовательностей метагеномов, не полагаясь на эталонные геномы.

Секвенирование метагеномов методом дробовика стало методом выбора для изучения и классификации микроорганизмов в различных экологических средах. Благодаря последним достижениям в технологии полногеномного секвенирования и постоянному улучшению качества и экономической эффективности крупномасштабное секвенирование становится все проще и быстрее. Это привело к значительному увеличению объема данных метагеномного секвенирования за последние несколько лет, что сделало их незаменимым ресурсом для изучения микробной темной материи. Для выяснения генетического состава метагеномных образцов обычно существует два основных подхода, каждый из которых имеет свои преимущества и недостатки. При первом подходе считывания секвенирования точно сопоставляются с известным аннотированным набором эталонных последовательностей генома, что позволяет быстро понять наличие известных организмов, генов и потенциальных функций. Во втором методе ряд базовой информации, полученной путем секвенирования, может быть собран в непрерывные фрагменты ДНК, называемые контигами или каркасами, посредством сборки de novo (без ссылки на геном). Этот процесс сборки может раскрыть нам информацию о некоторых ранее неописанных вещах. Микроорганизмы и их генетический состав. Однако оба метода страдают от одного и того же ограничения в функциональной аннотации генов, которая основана на функции прогнозирования в базах данных белков посредством поиска гомологии. Поэтому гены, которые не соответствуют семейству эталонных белков, обычно игнорируются и исключаются из последующих сравнительных анализов. Чтобы исключить зависимость от справочных наборов данных для оценки широты неисследованного функционального разнообразия (которое авторы называют функциональной темной материей), необходимы комплексные метагеномные сравнения. Но эта задача требует значительных вычислительных ресурсов, и достижение такой масштабируемости остается технически сложной задачей. Несмотря на некоторые превосходные недавние усилия по решению этой проблемы, метагеномы еще не были всесторонне исследованы для выявления функциональной темной материи. Здесь авторы представляют масштабируемый вычислительный подход для идентификации и характеристики функциональной темной материи, обнаруженной в метагеномах. Авторы впервые идентифицировали новые белковые пространства в 26 931 наборе метагеномных данных IMG/M, удалив все гены, которые соответствовали более чем 100 000 эталонным геномам или Pfam в базе данных IMG. Затем оставшиеся последовательности были сгруппированы в семейства белков, исследовано их таксономическое и экологическое распределение и, где это возможно, предсказана их третичная (трехмерная (3D)) структура.
Новое пространство последовательностей белков
Во-первых, авторы собрали все белковые последовательности длиной более 35 аминокислотных остатков из всех общедоступных эталонных геномов, метагеномов и метатранскриптомов на платформе IMG/M. В общей сложности все белковые последовательности были извлечены из 89 412 бактериальных геномов, 9 202 вирусных геномов, 3 073 геномов архей и 804 геномов эукариот, в результате чего в окончательный набор данных вошли в общей сложности 94 672 003 последовательности. В число эталонных геномов, включенных в исследование, вошли только изолированные геномы, за исключением метагеномов и индивидуально амплифицированных геномов. Аналогичным образом, для неклассифицированных метагеномов авторы извлекли все предсказанные белковые последовательности из каркасов длиной не менее 500 п.н. и длиной не менее 35 аминокислот, в общей сложности из 26 931 набора данных (20 759 метагеномов и 6 172 группы метатранскриптов), которые авторы называют Набор экологических данных (ED). В результате был получен неизбыточный набор данных, включающий в общей сложности 8 364 611 943 предсказанных белка или фрагмента белка. Чтобы идентифицировать функциональные компоненты темной материи в этом наборе данных, авторы сначала отбросили любые белковые последовательности, которые соответствовали последовательностям в базе данных Pfam или последовательностям в эталонном наборе геномов. Наконец, неизбыточный каталог, представляющий неизведанное пространство метагеномных белков, включает 1 171 974 849 белковых последовательностей (14% от общего числа).
новое семейство белков
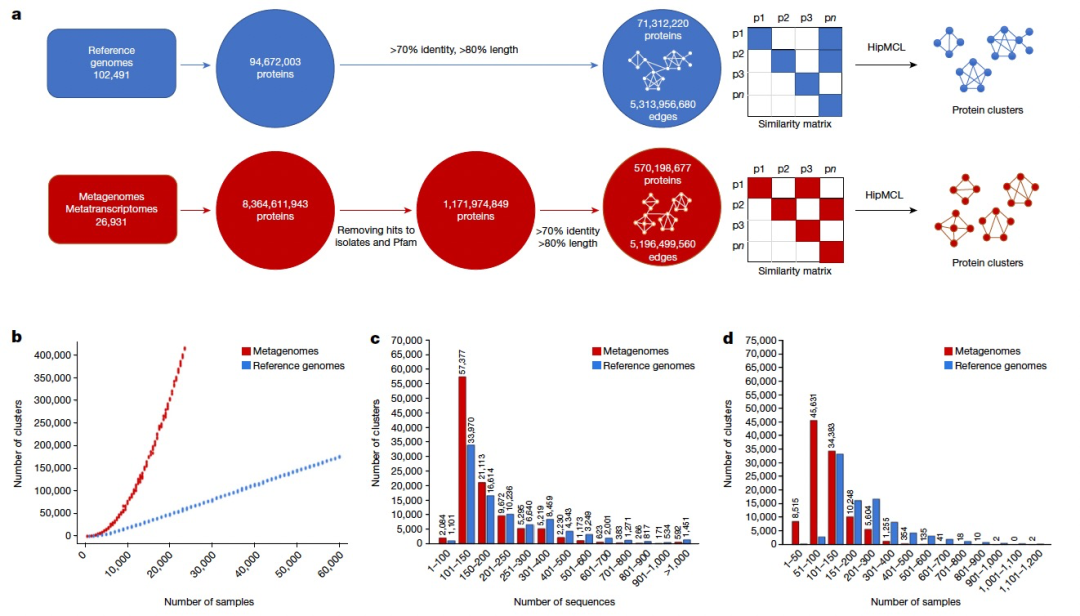
Рисунок 1
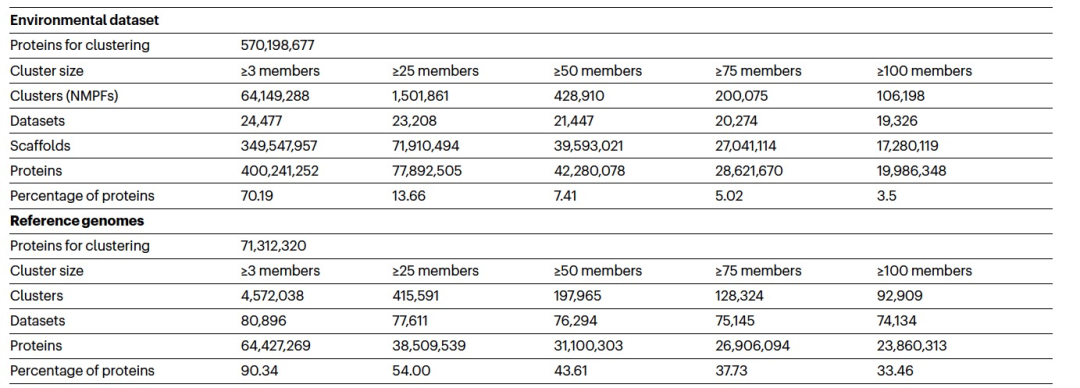
Таблица 1
Затем авторы кластеризовали 1,1 миллиарда белков ED. Для сравнения авторы также использовали тот же подход к 94 миллионам белков из эталонного генома. Сначала была построена матрица общего сходства для каждого из двух каталогов генов (т. е. белков из эталонного генома и белков из ED) путем подсчета всех значимых пар сходства последовательностей. Затем два графика были проанализированы для идентификации кластеров белков на основе сходства последовательностей. Для этой цели авторы использовали HipMCL — реализацию оригинального алгоритма MCL с массовым параллелизмом. Весь процесс от извлечения данных до создания кластера показан на рисунке 1a.
Хотя кластеры, насчитывающие не менее 50 членов (и, возможно, даже кластеры, насчитывающие не менее 25 членов), могут представлять собой потенциально функционально важные кластеры, авторы ограничили последующий анализ более крупными семьями, насчитывающими не менее 100 членов, чтобы сосредоточиться на большем количестве высококачественных данных. лучшие кандидаты на предсказанные структуры (табл. 1). Всего авторы выделили 106 198 семейств, насчитывающих не менее 100 членов, которые будем называть новыми семействами метагеномных белков (NMPF) (табл. 1 (правый столбец)). Для сравнения авторы идентифицировали 92 909 белковых кластеров в соответствующих белковых кластерах эталонного генома. Непосредственно сравнивая эти два набора кластеров (эталонные кластеры и кластеры белков ED), можно заметить, что для кластеров, состоящих как минимум из 3 членов, наблюдается увеличение более чем в 14 раз для кластеров белков ED и для кластеров, состоящих как минимум из 3 членов. 25 членов, наблюдается рост на 3 Более чем в два раза для кластеров с числом не менее 50 и 75 участников наблюдался рост примерно в 2 раза, для кластеров с числом не менее 100 членов также наблюдался рост (табл. 1); . Хотя пространство метагеномных последовательностей по своей природе более фрагментировано и имеет более высокую долю неправильных или неполных генов, чем эталонные геномы, эти результаты также демонстрируют, что еще остается много пространства белковых последовательностей для изучения. Это также подтверждается кривой разреженности, генерируемой кластерами, насчитывающими не менее 100 членов (рис. 1б). Эти кривые показывают, что с увеличением количества образцов количество кластеров увеличивается линейно для эталонного генома, но количество кластеров для метагенома увеличивается экспоненциально, не выходя на плато.
Распространение экосистемы
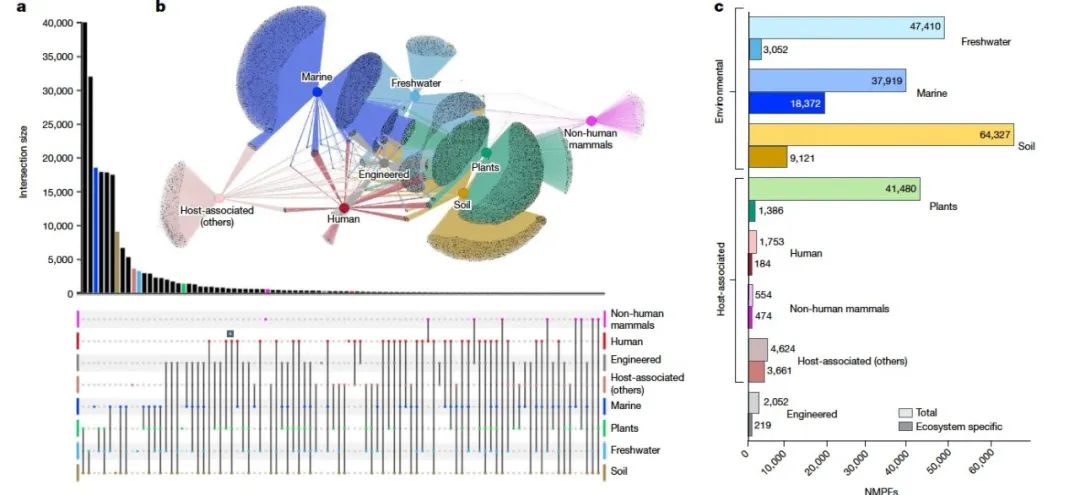
Рисунок 2
Определить Распространение НМПФ. экосистемы, автор использует GOLDбазу через платформу IMG/M данных Схема классификации экосистем собирает соответствующие элементы для каждого образца.данные。NMPFsРаспределение экосистемы следующее:картина2a,б. Три основные экосистемы GOLD здесь (экологическая, связанная с хозяином и инженерная) далее делятся на восемь более конкретных типов экосистем: пресноводная, морская, почвенная, растительная, человеческая, нечеловеческая млекопитающая, другие связанные с хозяином и искусственно созданная. Наблюдая за топологией сети,Встречается в трех обширных экосистемах.,Каждый NMPF имеет наименьшее количество общих генов.,Это согласуется с недавними наблюдениями за семействами белков из 13 174 метагеномов.,За исключением почвенно-растительных ассоциаций. Наибольшее количество NMPF распределено между почвой и растительной средой (62% почв и 96% семейств растений).,Это как и ожидалось,Потому что перекрытие выборки этих экосистем велико (картина2а). Далее следуют NMPF, общие для почвы и пресной воды.,В первую очередь это связано с тем, что образцы водно-болотных угодий и отложений классифицируются как пресноводные экосистемы. та же причина,Значительное дублирование также можно наблюдать между растительными и пресноводными NMPF, а также между почвенными, пресноводными и растительными NMPF. Напротив,Только 37% распределяются между пресноводными и морскими НПМП.,Белковые семейства менее распространены между людьми, млекопитающими, отличными от человека, и типами экосистем, связанных с хозяином. с другой стороны,Значительное перекрытие NMPF наблюдалось между человеческой и инженерной средой (картина2). это неудивительно,Учтите, что инженерная среда в основном содержит образцы экосистем, связанных с отходами жизнедеятельности человека (такими как твердые отходы и сточные воды). Сходным образом,Существует также совпадение между пресноводной и искусственно созданной средой, а также между пресноводными и связанными с хозяином типами (человек, млекопитающие, не относящиеся к человеку, и другие виды, связанные с хозяином). Эти совпадения могут указывать на явление,Например, фекальное загрязнение пресноводной среды.,Это приводит к сосуществованию одних и тех же NMPF и, следовательно, одних и тех же микробных сообществ в разных типах экосистем.
таксономическое распределение
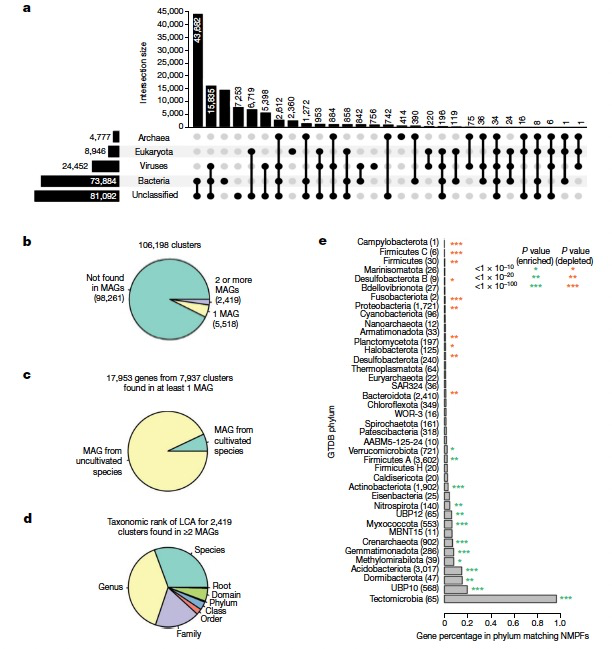
Рисунок 3
Автор на основе IMGбаза Таксономические назначения были сделаны для членов NMPF в каждом кластере на основе доступной таксономической информации для соответствующих каркасов в данных. В отсутствие таких аннотаций авторы использовали комбинацию других методов, чтобы определить таксономию каркаса. Из общего числа 17 280 119 каркасов IMG/M, содержащих членов NMPF, 8 049 154 были классифицированы как бактериальные, 382 761 как архейные, 1 184 393 как эукариотические, 1 406 588 как вирусные, а остальные 6 257 223 были неклассифицированными. Основываясь на соответствующем таксономическом назначении каркаса, таксономическое значение NMPF распределение, как показано на рисунке 3а. Большинство семейств белков включают последовательности с множественными таксономическими назначениями (например, бактериальные и неклассифицированные или бактериальные и вирусные). Самая большая категория включает семейства с бактериальными/неклассифицированными последовательностями, за которыми следуют вирусные/неклассифицированные и бактериальные/вирусные последовательности. Небольшая группа семейств отнесена к эукариотам, еще меньшая группа — к археям. Наконец, было 7253 кластера вообще без таксономической информации. Далее авторы оценили различия между недавно выявленным микробиомом Земли (геномами). from Earth's В каталоге микробиомов (GEM) обнаружены ли какие-либо белки NMPF (и соответствующие им семейства)? В частности, авторы исследовали, были ли каркасы, содержащие гены NMPF, распределены среди 52 515 MAG в каталоге GEM. Результаты показали, что в каталоге GEM было обнаружено 17 953 гена, происходящих из 7 937 NMPF (7,4% от общего числа) (рис. 3б, в), причем подавляющее большинство (93%) происходит от некультивируемых видов. Для тех семейств, которые встречаются в двух или более MAG, можно отметить, что они имеют сильно узкое таксономическое распространение: более двух третей семейств ограничены одним видом или родом, и лишь очень небольшое количество семейств распределены по нескольким семействам, отрядам или типам (рис. 3г). Было обнаружено, что NMPF обогащен несколькими типами, обычно встречающимися в почвенной среде (например, Gemmatimonadota, Acidobacteriota, Crenarchaeota и Myxococcota), и менее распространен в нескольких типах, обнаруженных в средах, связанных с людьми и другими хозяевами (Firmicutes, Proteobacteria и Bacteroidota; рисунок 3e). ). В совокупности эти результаты показывают, что, несмотря на улучшение общих процессов и крупномасштабные реконструкции MAG, значительная часть функционального разнообразия остается таксономически изолированной.
Распространение метаданных
Далее авторы изучили географическое распределение НМПФ. Очень немногие семьи (1,372 человека, или 1,3%), имели ограниченное географическое распространение (в пределах 1 км), когда максимально допустимое расстояние составляло 1 км.,000 км, это число увеличивается лишь незначительно (4,330, что составляет 4%). Большинство этих семейств встречается в растениях, почве и пресноводных экосистемах. Очень немногие из этих семейств включают представителей, обнаруженных в морских экосистемах или образцах человека, что соответствует более высокому распространению микробов в этих экосистемах. Большинство НПФ (64,186 или 60,44%) состояли из смеси белков метагенома и метатранскриптома, что еще раз подтверждало их существование, а 38,292 (36,06%) NMPF содержали белки, обнаруженные только в метагеномах, 3,720 (3,50%) NMPF содержали белки, обнаруженные только в метатранскриптоме. По мере уменьшения числа членов семьи постепенно снижается процент семей, содержащих представителей метагенома и метатранскриптома. В то же время эти NMPF, обнаруженные как в метагеномах, так и в метатранскриптомах, также имели самое широкое выборочное распространение, то есть эти семейства были обнаружены в большинстве образцов. Большинство этих семейств классифицируются как экологические экосистемы (в основном почвы и, в меньшей степени, образцы морской и пресноводной воды) и содержат в основном бактерии и неклассифицированные последовательности. Чтобы оценить распределение белков нового семейства в данных секвенирования окружающей среды, авторы сравнили количество новых белков, извлеченных из каждого каркаса и использованных в этом исследовании, с общим количеством генов/белков в соответствующем каркасе. Большинство проанализированных каркасов (13,407,728, что составляет 77,59%), содержат как новые, так и известные гены. Сравнение количества новых и общего количества генов в этих каркасах не выявило корреляции между размером каркаса или общим количеством генов на каркас и количеством новых генов. Самые большие каркасы в исследовании авторов (4,302гены)содержит только новую последовательность。Обычно,Самые крупные исследованные каркасы содержали лишь ограниченное количество новых последовательностей.,и получены из бактериальных или неклассифицированных метагеномных образцов. Напротив,Длина (и количество генов) каркасов, содержащих большинство новых последовательностей, сильно различалась.,И большинство из них происходят от вирусов.
Структурное распределение
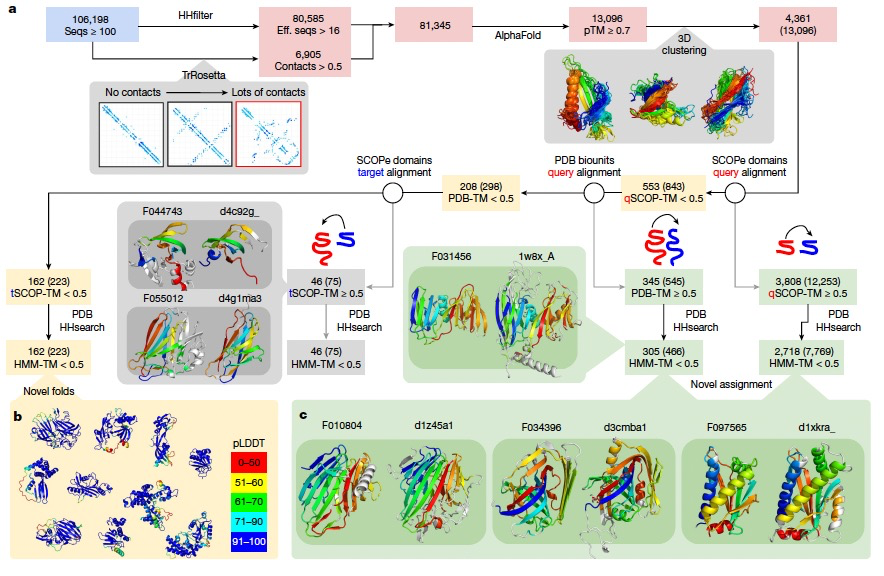
Рисунок 4
Недавние прорывы в предсказании структуры белков позволили быстро и точно определить структурные характеристики белковых последовательностей. Показано, что метагеномные последовательности являются особенно богатым источником для открытия новых структур. Здесь авторы использовали AlphaFold2 для анализа NMPF, содержащих по меньшей мере 16 различных последовательностей или хорошо структурированных белков, предсказанных TrRosetta. Краткое изложение результатов показано на рисунке 4а. Среди 81 345 NMPF, отвечающих вышеуказанным условиям, было спрогнозировано 80 585 3D-моделей, из которых 13 096 NMPF имели высокую достоверность прогнозирования (прогнозируемый балл TM (pTM) > 0,700). Эти прогнозы с высокой степенью достоверности, основанные на структурной кластеризации, представляют собой 4361 уникальную структуру. Чтобы проверить новизну или функциональность этих структур, авторы сравнили их с экспериментально определенными структурами в SCOP-Extended (SCOPe) и комплексами в PDB. В общей сложности 3808 структур (содержащих 12 253 NMPF) имеют значительное структурное перекрытие (оценка TM) как минимум с 1 доменом SCOPe. > 0,5). Из них 2718 (в том числе 7769 NMPF) имели высокие совпадения, что указывает на то, что 62,3% высококачественных прогнозов имели хотя бы некоторое сходство с доменом SCOPe или комплексом в PDB. Эти новые назначения, основанные на структурном сходстве, теперь можно использовать для прогнозирования функции соответствующих последовательностей. На рисунке 4c показано несколько примеров. Например, семейство F034396 не имело совпадений при сопоставлении с PDB с помощью HHsearch, но имело сильное совпадение при сопоставлении с доменом SCOPe с использованием структурного поиска. Авторы подчеркивают, что эти сценарии следует рассматривать как обоснованные предсказания, которые необходимо проверить экспериментально, поскольку одни и те же складки не всегда соответствуют одним и тем же функциям. Однако некоторая проверка и дополнительная функциональная аннотация могут быть выполнены путем объединения этих новых назначений с другими метаданными NMPF, такими как генетический симбиоз. Чтобы подтвердить, что оставшиеся 553 белка без совпадений SCOPe представляют собой новые складки, авторы провели более исчерпывающий поиск комплексов во всех организмах PDB, включая все возможные варианты расположения цепей. В общей сложности 345 моделей попали как минимум в одну запись PDB, 305 из которых представляли собой дополнительные новые назначения. Остальные 208 были обработаны для дальнейшей фильтрации, в результате чего было удалено 50% прогнозов, структура которых соответствовала домену SCOPe. В конечном итоге, 162 складки и/или ориентации домен-домен были идентифицированы из 223 NMPF, которые считались новыми (рис. 4b).
Ссылки
Pavlopoulos, G.A., Baltoumas, F.A., Liu, S. et al. Unraveling the functional dark matter through global metagenomics. Nature 622, 594–602 (2023).
https://doi.org/10.1038/s41586-023-06583-7



Неразрушающее увеличение изображений одним щелчком мыши, чтобы сделать их более четкими артефактами искусственного интеллекта, включая руководства по установке и использованию.
Копикодер: этот инструмент отлично работает с Cursor, Bolt и V0! Предоставьте более качественные подсказки для разработки интерфейса (создание навигационного веб-сайта с использованием искусственного интеллекта).
Новый бесплатный RooCline превосходит Cline v3.1? ! Быстрее, умнее и лучше вилка Cline! (Независимое программирование AI, порог 0)

Разработав более 10 проектов с помощью Cursor, я собрал 10 примеров и 60 подсказок.
Я потратил 72 часа на изучение курсорных агентов, и вот неоспоримые факты, которыми я должен поделиться!

Идеальная интеграция Cursor и DeepSeek API
DeepSeek V3 снижает затраты на обучение больших моделей
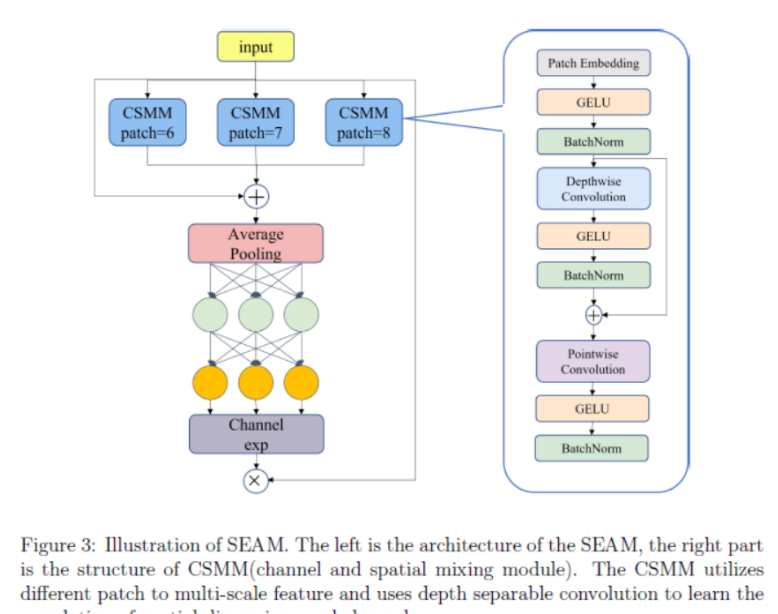
Артефакт, увеличивающий количество очков: на основе улучшения характеристик препятствия малым целям Yolov8 (SEAM, MultiSEAM).

DeepSeek V3 раскручивался уже три дня. Сегодня я попробовал самопровозглашенную модель «ChatGPT».

Open Devin — инженер-программист искусственного интеллекта с открытым исходным кодом, который меньше программирует и больше создает.
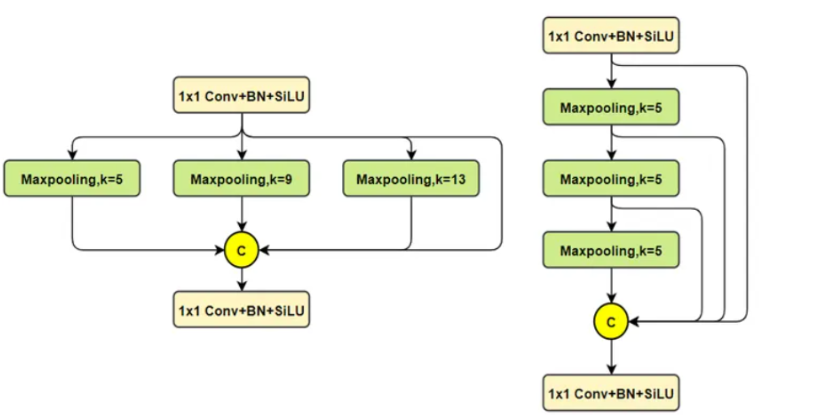
Эксклюзивное оригинальное улучшение YOLOv8: собственная разработка SPPF | SPPF сочетается с воспринимаемой большой сверткой ядра UniRepLK, а свертка с большим ядром + без расширения улучшает восприимчивое поле

Популярное и подробное объяснение DeepSeek-V3: от его появления до преимуществ и сравнения с GPT-4o.

9 основных словесных инструкций по доработке академических работ с помощью ChatGPT, эффективных и практичных, которые стоит собрать
Вызовите deepseek в vscode для реализации программирования с помощью искусственного интеллекта.
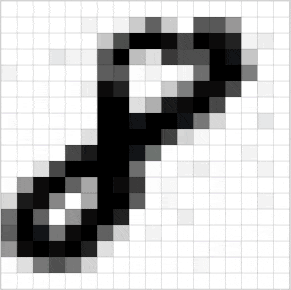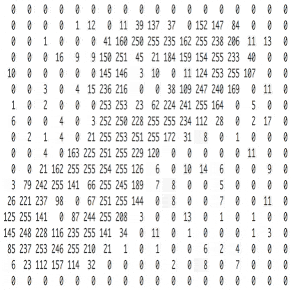
Познакомьтесь с принципами сверточных нейронных сетей (CNN) в одной статье (суперподробно)
50,3 тыс. звезд! Immich: автономное решение для резервного копирования фотографий и видео, которое экономит деньги и избавляет от беспокойства.
Cloud Native|Практика: установка Dashbaord для K8s, графика неплохая
Краткий обзор статьи — использование синтетических данных при обучении больших моделей и оптимизации производительности
MiniPerplx: новая поисковая система искусственного интеллекта с открытым исходным кодом, спонсируемая xAI и Vercel.
Конструкция сервиса Synology Drive сочетает проникновение в интрасеть и синхронизацию папок заметок Obsidian в облаке.
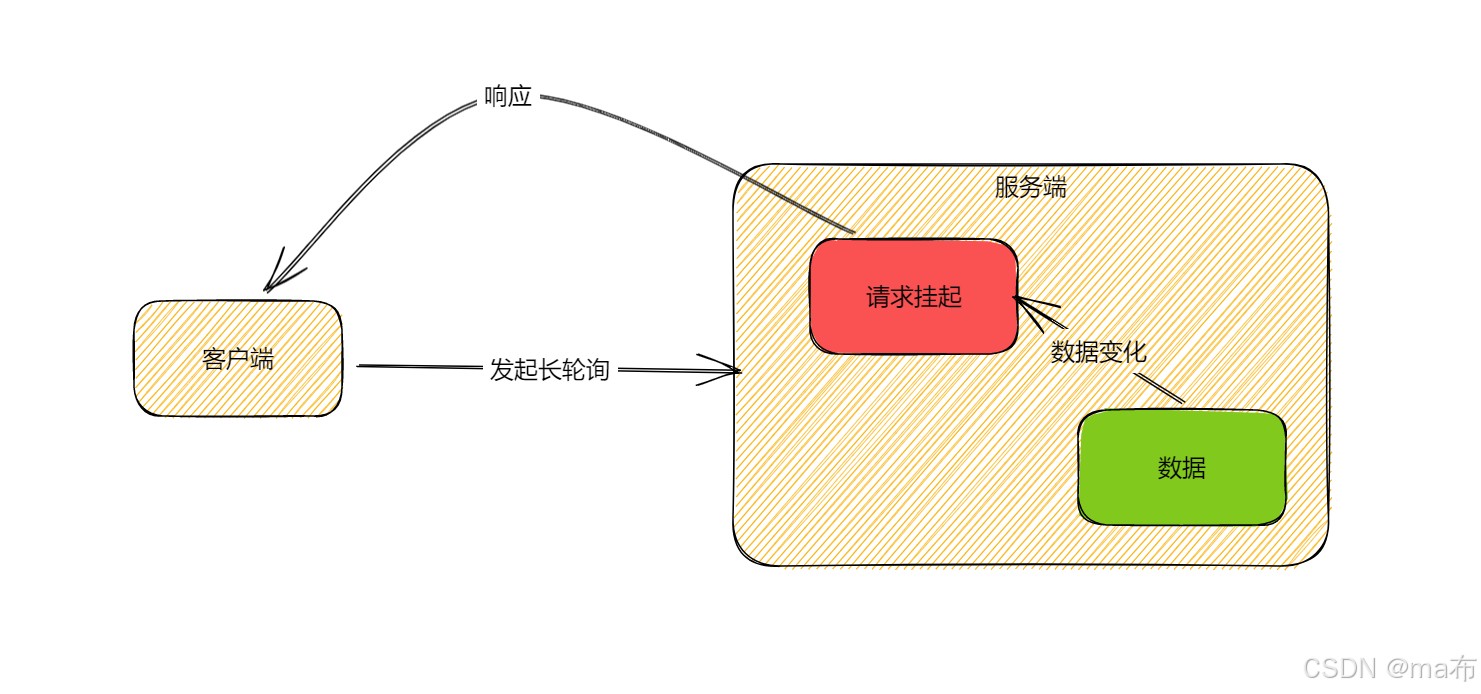
Центр конфигурации————Накос
Начинаем с нуля при разработке в облаке Copilot: начать разработку с минимальным использованием кода стало проще

[Серия Docker] Docker создает мультиплатформенные образы: практика архитектуры Arm64

Обновление новых возможностей coze | Я использовал coze для создания апплета помощника по исправлению домашних заданий по математике

Советы по развертыванию Nginx: практическое создание статических веб-сайтов на облачных серверах
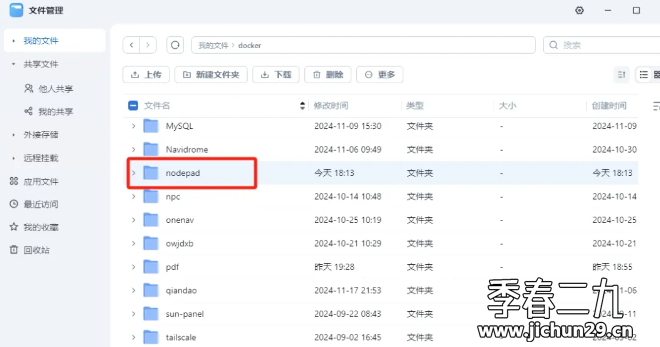
Feiniu fnos использует Docker для развертывания личного блокнота Notepad

Сверточная нейронная сеть VGG реализует классификацию изображений Cifar10 — практический опыт Pytorch
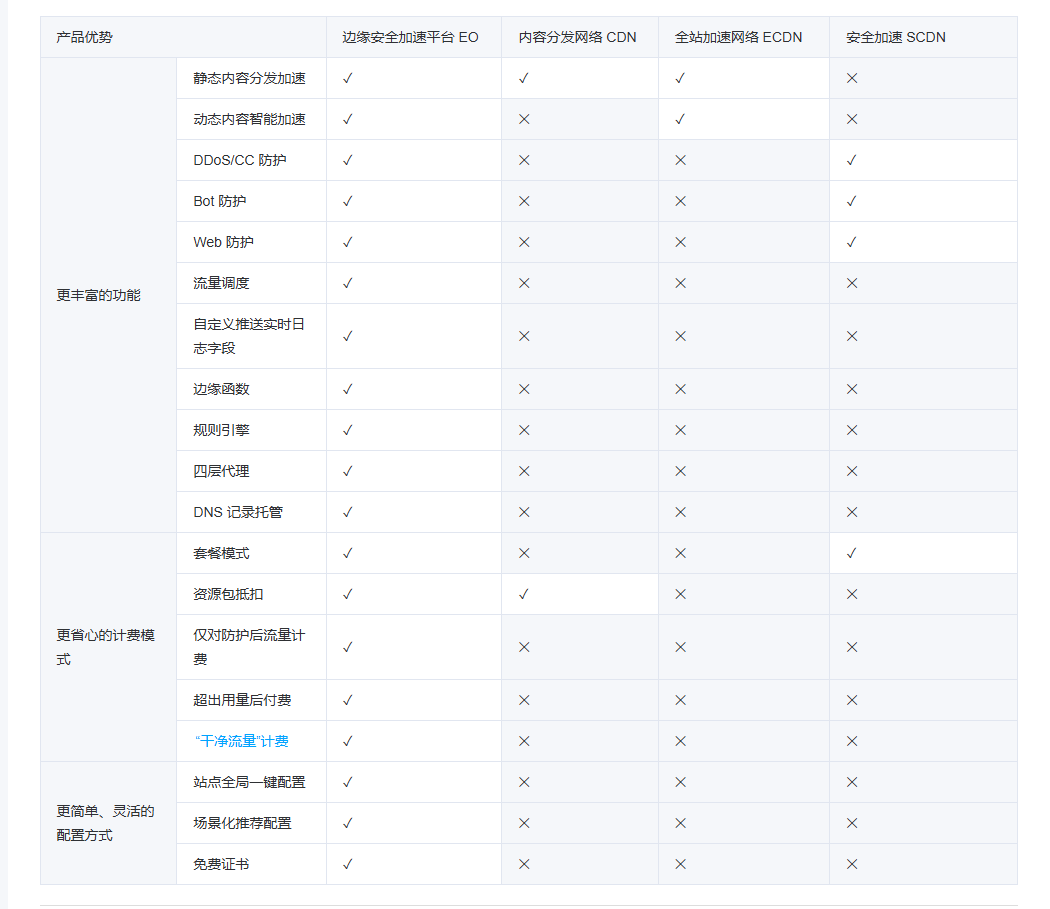
Начало работы с EdgeonePages — новым недорогим решением для хостинга веб-сайтов
[Зона легкого облачного игрового сервера] Управление игровыми архивами