Повторное появление набора данных об отдельных клетках рака желудка GSE163558 (1): загрузка данных, сортировка и считывание
Делиться — это отношение

Предисловие
Привет друзья,Я маленький ученик дерева навыков Шэнсинь «Я не ем яичные желтки». в следующий период времени,Я начну новую серию статей по обмену опытом,всемсистема Организация кода для секвенирования отдельных клеток。Эта серия включает, помимо прочего, следующие:Загрузка и считывание данных, контроль качества и групповое удаление, групповой дифференциальный анализ;。
Он в основном охватывает все методы рутинного анализа секвенирования отдельных клеток и особенно подходит для новичков и начинающих изучать систему. Поэтому приглашаем всех продолжать следить за обновлениями, подписываться на официальный аккаунт, ставить лайки + комментировать + собирать + смотреть. Ваша поддержка будет моей мотивацией к обновлению. Заранее всем спасибо.
1. Знакомство с литературой
Название документа, воспроизведенного в этой серии, — «Выявление транскрипционной гетерогенности органоспецифических метастазов при раке желудка человека с использованием секвенирования одноклеточной РНК». Автором-корреспондентом является профессор Фань Сяохуэй из Чжэцзянского университета, статья была опубликована в журнале Clin Transl Med в 2022 году (IF=10,6).

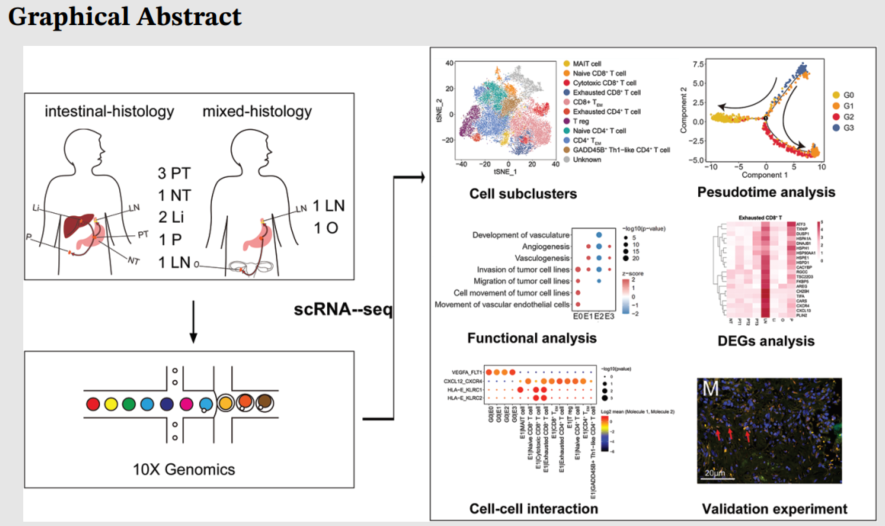
краткое содержание:
Цель: Выявление биологических особенностей рака желудка (РЖ) и его метастазов посредством анализа секвенирования отдельных клеток.
метод: В основном для технологии одноклеточного секвенирования от 6 пациентов было собрано 10 образцов свежих тканей (включая первичные опухоли, прилегающие ткани и метастазы различных органов и тканей) от 6 пациентов. и подтверждено с использованием гистологического анализа и наборов данных массовой транскрипции.
результат:
- Злокачественные эпителиальные субпопуляционные клетки и характеристики инвазии、Склонность к внутрибрюшинным метастазы, эпителиально-мезенхимальный переход, индуцированный фенотипом раковых стволовых клеток или характеристиками, подобными состоянию покоя. Результаты анализа выживаемости КМ на основе TCGA рака желудка, в свою очередь, показывают,Первые три гена, относящиеся к подгруппе злокачественного эпителия, являются факторами риска для прогноза пациентов с РЖ.
- Иммунные и стромальные клетки проявляют клеточную гетерогенность и создают проопухолевое и иммуносупрессивное микроокружение.
- Сигнатура из 20 генов, основанная на истощенных CD8+ Т-клетках лимфоидного происхождения, может предсказать лимфатические метастазы рака желудка, и этот результат был подтвержден в TCGAочереди.
- Помимо злокачественных опухолевых клеток, в качестве возможных были идентифицированы субпопуляции эндотелиальных клеток, инвариантных Т-клеток, ассоциированных со слизистой оболочкой, Т-клеточноподобных В-клеток, плазмацитоидных дендритных клеток, макрофагов, моноцитов и нейтрофилов. Помогают взаимодействовать с цитотоксическими/истощенными CD8+. Т-клетки и/или естественные клетки-киллеры (NK) (HLA-E-KLRC1/KLRC2), NKG2A (KLRC1) могут стать новой мишенью для лечения рака желудка.
- Экспрессия PD-1 в CD8+ Т-клетках предсказывает клинический ответ на ингибиторы PD-1 у пациентов с раком желудка.
в заключение: Это исследование обеспечивает глубокое понимание гетерогенного микроокружения первичных опухолей и органоспецифических метастазов рака желудка, обеспечивая точную диагностику и лечение.
Выше приведено введение к этой статье. Далее мы входим в часть анализа данных и начинаем загрузку и чтение данных.
2. Загрузка и сортировка данных.
Данные статьи о секвенировании отдельных клеток хранятся в базе данных GEO под номером GSE163558 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE163558). Этот набор данных содержит в общей сложности 10 образцов свежих тканей от 6 пациентов с раком желудка (включая первичные опухоли, прилегающие ткани и метастазы из различных органов и тканей):
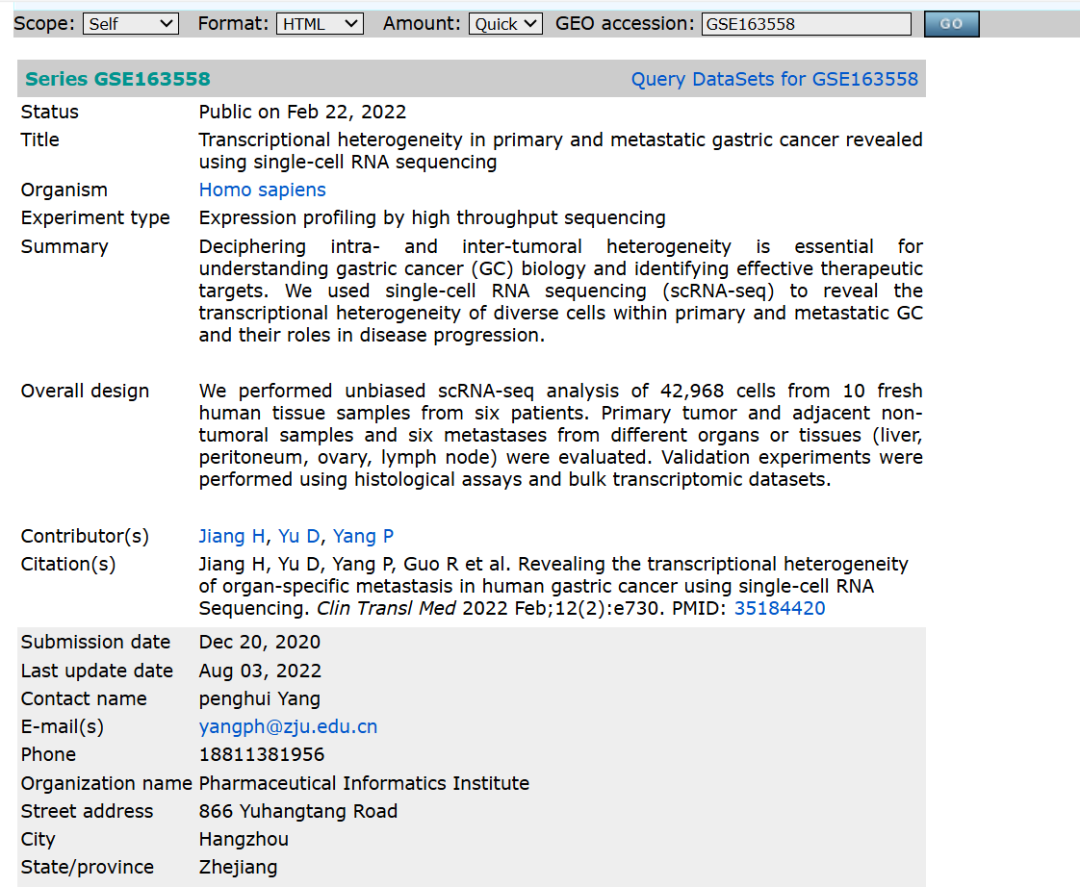
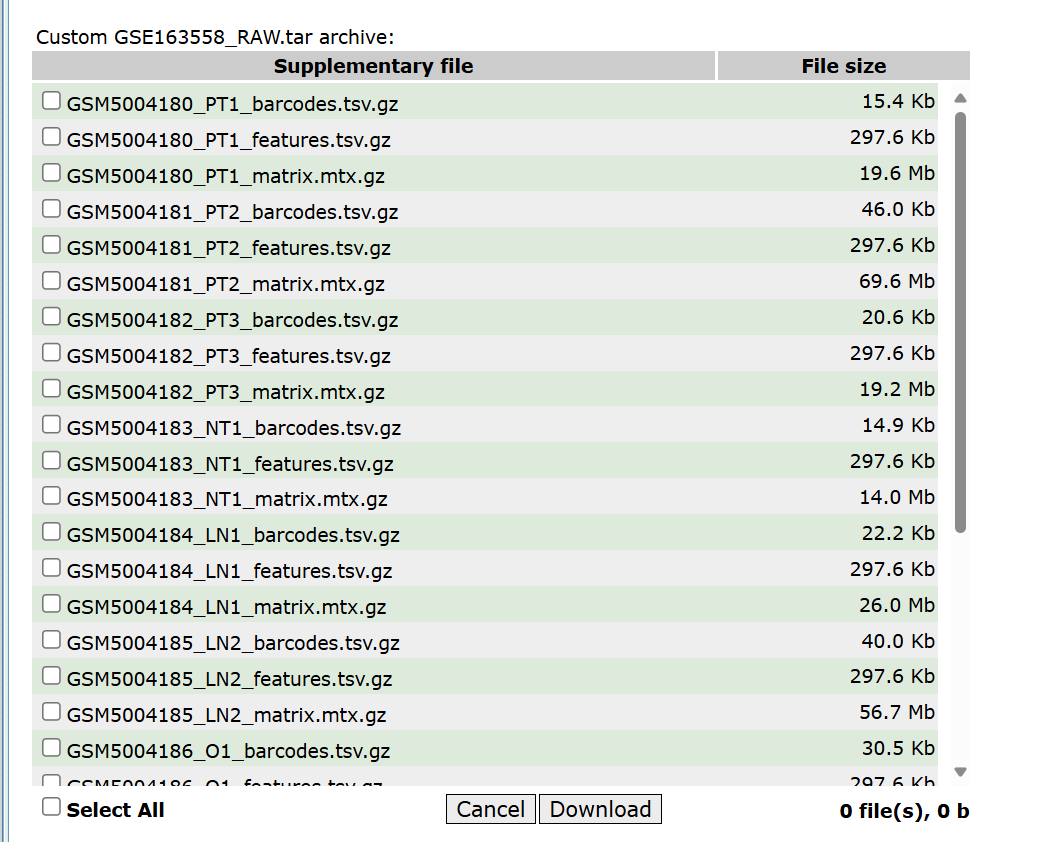
Как видите, формат файла данных — стандартный файл 10X. Стандартный файл 10X содержит три файла: barcodes.tsv.gz, Features.tsv.gz и Matrix.mtx.gz, созданные в результате сравнительного анализа Cellranger, которые соответственно представляют метку клетки (штрих-код), идентификатор гена (функция), данные экспрессии ( матрица).
Прежде чем читать файл, нам нужно загрузить пакет R:
rm(list=ls())
options(stringsAsFactors = F)
library(Seurat)
library(data.table)
library(dplyr)
После загрузки данных начните обработку исходных файлов, организуйте исходные файлы в barcodes.tsv.gz, Features.tsv.gz и matrix.mtx.gz и поместите их в соответствующие папки. Код выглядит следующим образом:
setwd("")
dir='GSE163558_RAW/'
fs=list.files('GSE163558_RAW/','^GSM')
fs
library(tidyverse)
samples=str_split(fs,'_',simplify = T)[,1]
##Обработка данных и организация исходных файлов в barcodes.tsv.gz, Features.tsv.gz и Matrix.mtx.gz в соответствующих папках.
#Изменяем имена файлов пакетно на Функция Read10X() распознала имена
lapply(unique(samples),function(x){
# x = unique(samples)[1]
y=fs[grepl(x,fs)]
folder=paste0("GSE163558_RAW/", paste(str_split(y[1],'_',simplify = T)[,1:2], collapse = "_"))
dir.create(folder,recursive = T)
#Создаем подпапки для каждого образца
file.rename(paste0("GSE163558_RAW/",y[1]),file.path(folder,"barcodes.tsv.gz"))
#Переименовываем файл и перемещаем его в соответствующую подпапку
file.rename(paste0("GSE163558_RAW/",y[2]),file.path(folder,"features.tsv.gz"))
file.rename(paste0("GSE163558_RAW/",y[3]),file.path(folder,"matrix.mtx.gz"))
})
3. Чтение и объединение данных
После сортировки стандартных файлов 10X используйте функцию Read10X() для объединения трех стандартных файлов и получения разреженной матрицы экспрессии (поведенческие гены, столбцы в виде ячеек, формат dgCMatrix). На основе разреженной матрицы выражений «tmp» используйте функцию CreateSeuratObject для создания объекта Seurat. Для нескольких образцов требуется пакетное чтение нескольких файлов. Здесь мы используем функцию lapply (вы также можете использовать цикл for).
dir='GSE163558_RAW/'
samples=list.files( dir )
samples
sceList = lapply(samples,function(pro){
# pro=samples[1]
print(pro)
tmp = Read10X(file.path(dir,pro ))
if(length(tmp)==2){
ct = tmp[[1]]
}else{ct = tmp}
sce =CreateSeuratObject(counts = ct ,
project = pro ,
min.cells = 5,
min.features = 300 )
return(sce)#Верните созданный объект Seurat и сохраните его в sceList.
})
View(sceList)
Полученный здесь sceList на самом деле содержит объекты Сёра из 10 образцов:
Посмотрите один из них:
PT1 <- sceList[1]
View(PT1)
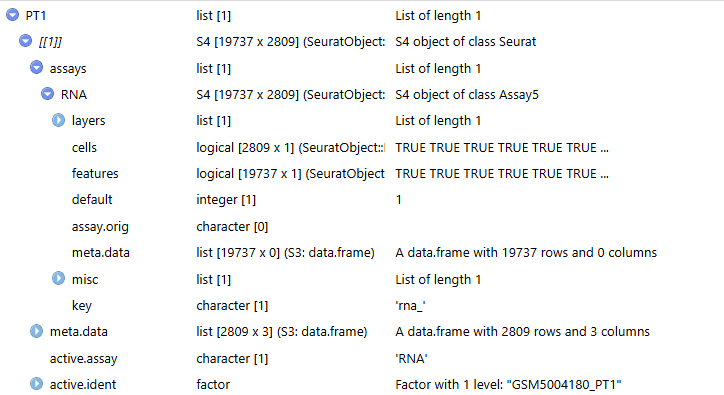
Как видите, это Сёра, содержащий различные элементы. V5объект(V5Версияизassaysобъектесть еще нижеlayersизструктура)(Подробности смотрите в предыдущем твитеhttps://mp.weixin.qq.com/s/2dtIS1qd0tPM1dQRCKptAQ)。
Далее нам нужно использовать функцию слияния пакета Seurat, чтобы объединить десять Seurat в один объект Seurat.
do.call(rbind,lapply(sceList, dim))
sce.all=merge(x=sceList[[1]],
y=sceList[ -1 ],
add.cell.ids = samples )
names(sce.all@assays$RNA@layers)
На данный момент, хотя мы завершили создание объекта Сёра, вы можете видеть, что для десяти образцов еще осталось 10 слоев. Если дальнейшей обработки не будет, данные при последующем извлечении подсчетов будут неполными, и анализ будет продолжать давать ошибки. Поэтому нам нужно использовать функцию JoinLayers для объединения слоев.
sce.all[["RNA"]]$counts
# Alternate accessor function with the same result
LayerData(sce.all, assay = "RNA", layer = "counts")
#Посмотрите на изменения в сцене до и после слияния
sce.all
sce.all <- JoinLayers(sce.all)
sce.all
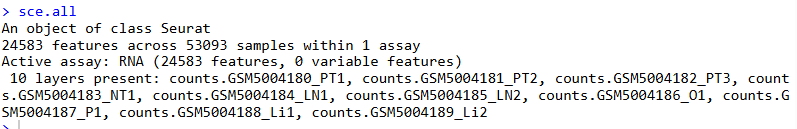
Это до интеграции:
Это после интеграции:
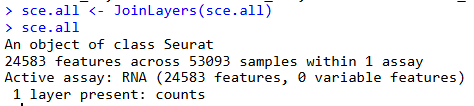
Видно, что после слияния Сёра и слоев наконец получается полный объект Сёра «sce.all». Мы можем проверить некоторую информацию внутри «sce.all», чтобы проверить, полны ли данные.
dim(sce.all[["RNA"]]$counts )
as.data.frame(sce.all@assays$RNA$counts[1:10, 1:2])
head(sce.all@meta.data, 10)
table(sce.all$orig.ident)
length(sce.all$orig.ident)
Мы можем просмотреть количество клеток на образец и общее количество клеток:

4. Добавьте информацию о группировке метаданных.
После успешного создания объекта Сёра «sce.all» нам также необходимо добавить в выборку информацию о группировке метаданных, чтобы облегчить последующие сравнения между различными группами и извлечение подгрупп для дальнейшего анализа. Сначала мы проверяем, какова существующая информация метаданных:
library(stringr)
phe = sce.all@meta.data
table(phe$orig.ident)
View(phe)
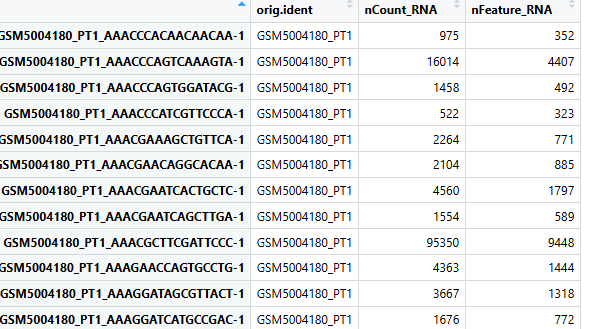
Как видите, единственная существующая информация: orig.ident (название образца), nCount_RNA (количество UMI на клетку) и nFeature_RNA (количество генов, обнаруженных в каждой клетке). Однако исходный текст содержит такую информацию, как источник пациента, источник ткани и место метастазирования образца. Эту информацию можно отличить по номеру образца. Поэтому мы можем использовать функции обработки текста «str_split» и «gsub» для обработки номера пациента и добавления указанной выше информации в метаданные.
Функция str_split используется для разделения строк:
phe$group = str_split(phe$orig.ident,'[_]',simplify = T)[,2]
Добавить информацию о группе мест метастазирования
phe$sample = phe$orig.ident
phe$sample = gsub("GSM\\d+_PT\\d+", "GC", phe$sample)
phe$sample = gsub("GSM\\d+_LN\\d+", "GC_lymph_metastasis", phe$sample)
phe$sample = gsub("GSM\\d+_O1", "GC_ovary_metastasis", phe$sample)
phe$sample = gsub("GSM\\d+_P1", "GC_peritoneum_metastasis", phe$sample)
phe$sample = gsub("GSM\\d+_Li\\d+", "GC_liver_metastasis", phe$sample)
phe$sample = gsub("GSM\\d+_NT\\d+", "adjacent_nontumor", phe$sample)
table(phe$sample)
Добавить информацию об источнике пациента
phe$patient = phe$orig.ident
table(phe$patient)
phe$patient = gsub("GSM5004180_PT1|GSM5004188_Li1", "Patient1", phe$patient)
phe$patient = gsub("GSM5004181_PT2|GSM5004183_NT1", "Patient2", phe$patient)
phe$patient = gsub("GSM5004186_O1", "Patient3", phe$patient)
phe$patient = gsub("GSM5004185_LN2", "Patient5", phe$patient)
phe$patient = gsub("GSM5004187_P1", "Patient6", phe$patient)
phe$patient = gsub("GSM\\S+", "Patient4", phe$patient)
table(phe$patient)
Также в статье есть группа, разделенная на обычные NT, исходные PT и перенесенные M, которые добавляются с помощью функции ifelse.
phe$tissue <- ifelse(phe$orig.ident %in% c("GSM5004180_PT1","GSM5004181_PT2","GSM5004182_PT3"),"PT",
ifelse(phe$orig.ident %in% c("GSM5004183_NT1"),"NT","M"))
table(phe$tissue)
sce.all@meta.data = phe
View(phe)
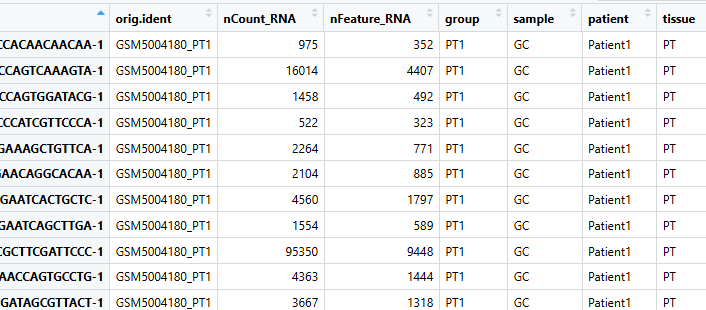
Наконец, вы можете видеть, что мы успешно добавили всю демонстрационную информацию. На этом этапе данные загружены, отсортированы и прочитаны, и можно начинать последующий стандартный процесс Seurat V5.
Заключение
В этом выпуске мы кратко рассмотрели аннотацию литературы, загрузили данные секвенирования отдельных клеток в формате 10X для 10 образцов набора данных о раке желудка GSE163558 и организовали файлы. После чтения файлов 10X в пакетном режиме они объединились и успешно сконструировали. объект Сера, на основе которого клиническая информация о пациенте добавляется к информации о группировке метаданных. В следующем выпуске мы будем следовать стандартному процессу Seurat V5 на этой основе, проводить контроль качества объектов Seurat, использовать гармоническую интеграцию для пакетной обработки, а также выполнять уменьшение размерности и кластеризацию в соответствии со стандартным процессом.



Неразрушающее увеличение изображений одним щелчком мыши, чтобы сделать их более четкими артефактами искусственного интеллекта, включая руководства по установке и использованию.
Копикодер: этот инструмент отлично работает с Cursor, Bolt и V0! Предоставьте более качественные подсказки для разработки интерфейса (создание навигационного веб-сайта с использованием искусственного интеллекта).
Новый бесплатный RooCline превосходит Cline v3.1? ! Быстрее, умнее и лучше вилка Cline! (Независимое программирование AI, порог 0)

Разработав более 10 проектов с помощью Cursor, я собрал 10 примеров и 60 подсказок.
Я потратил 72 часа на изучение курсорных агентов, и вот неоспоримые факты, которыми я должен поделиться!

Идеальная интеграция Cursor и DeepSeek API
DeepSeek V3 снижает затраты на обучение больших моделей
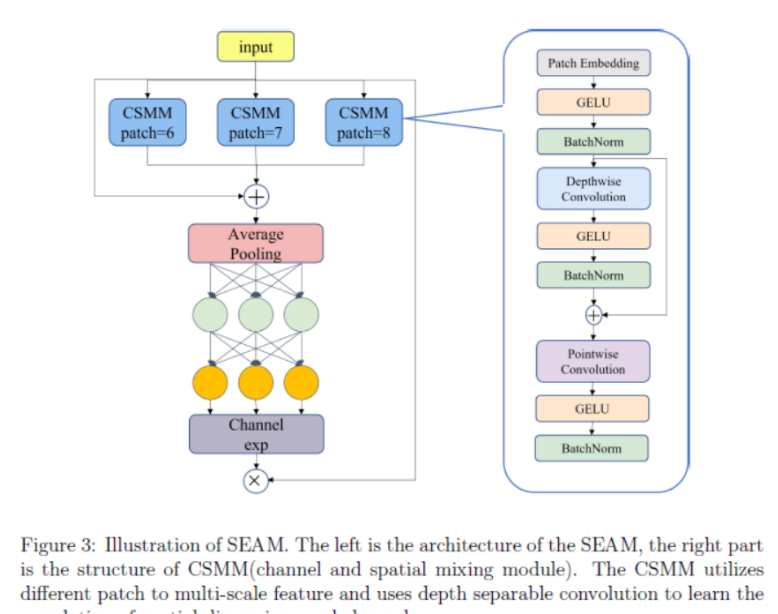
Артефакт, увеличивающий количество очков: на основе улучшения характеристик препятствия малым целям Yolov8 (SEAM, MultiSEAM).

DeepSeek V3 раскручивался уже три дня. Сегодня я попробовал самопровозглашенную модель «ChatGPT».

Open Devin — инженер-программист искусственного интеллекта с открытым исходным кодом, который меньше программирует и больше создает.
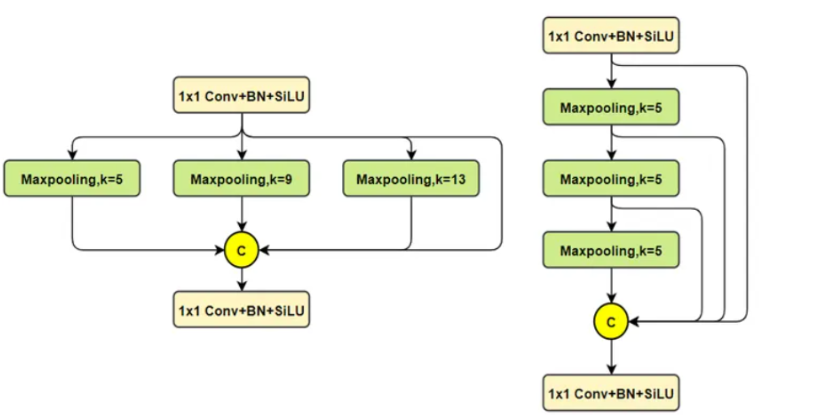
Эксклюзивное оригинальное улучшение YOLOv8: собственная разработка SPPF | SPPF сочетается с воспринимаемой большой сверткой ядра UniRepLK, а свертка с большим ядром + без расширения улучшает восприимчивое поле

Популярное и подробное объяснение DeepSeek-V3: от его появления до преимуществ и сравнения с GPT-4o.

9 основных словесных инструкций по доработке академических работ с помощью ChatGPT, эффективных и практичных, которые стоит собрать
Вызовите deepseek в vscode для реализации программирования с помощью искусственного интеллекта.
Познакомьтесь с принципами сверточных нейронных сетей (CNN) в одной статье (суперподробно)
50,3 тыс. звезд! Immich: автономное решение для резервного копирования фотографий и видео, которое экономит деньги и избавляет от беспокойства.
Cloud Native|Практика: установка Dashbaord для K8s, графика неплохая
Краткий обзор статьи — использование синтетических данных при обучении больших моделей и оптимизации производительности
MiniPerplx: новая поисковая система искусственного интеллекта с открытым исходным кодом, спонсируемая xAI и Vercel.
Конструкция сервиса Synology Drive сочетает проникновение в интрасеть и синхронизацию папок заметок Obsidian в облаке.
Центр конфигурации————Накос
Начинаем с нуля при разработке в облаке Copilot: начать разработку с минимальным использованием кода стало проще

[Серия Docker] Docker создает мультиплатформенные образы: практика архитектуры Arm64

Обновление новых возможностей coze | Я использовал coze для создания апплета помощника по исправлению домашних заданий по математике

Советы по развертыванию Nginx: практическое создание статических веб-сайтов на облачных серверах
Feiniu fnos использует Docker для развертывания личного блокнота Notepad

Сверточная нейронная сеть VGG реализует классификацию изображений Cifar10 — практический опыт Pytorch
Начало работы с EdgeonePages — новым недорогим решением для хостинга веб-сайтов
[Зона легкого облачного игрового сервера] Управление игровыми архивами