полный транскриптом | три поколенияполный транскриптоманалитический процесс (PacBio & ONT )-- Flair
Сегодня мы представляем инструмент, который использует три поколения полноразмерных данных транскриптов для коррекции транскриптов, кластеризации, анализа альтернативного сплайсинга, количественной оценки и дифференциального анализа. - FLAIR。из Калифорнийского университета в Санта-Крузе(University of California,Santa Cruz)изAngela Brooksкоманда(картина1)развиватьизполная переменная транскрипциякнига(isoform)Инструменты анализаFLAIR (Full-Length Alternative Isoform analysis of RNA),В2020Год03луна18№ опубликовано в《Nature Communications》в журнале,Название Full-length transcript characterization of SF3B1 mutation in chronic lymphocytic leukemia reveals downregulation of retained introns。Этот инструмент можно использоватьОпределите стенограммы с высокой степенью достоверности,Анализ событий дифференциального сдвигаиДифференциальный анализ изоформ (изоформ) транскриптов。

Зрелый предшественник м РНК (Пре-м РНК) изсращиваниеда Зависит отодин называетсясплайсосома(Spliceosome)изRNA-выполнение белкового комплексаиз。сплайсосома Зависит от 5 индивидуальный Маленькийизрибонуклеопротеиновые частицы(snRNPs,включать U1、U2、U4、U5 и U6) и нет Факторы snRNP собраны. здесь 5 индивидуальный мя РНП,U2 snRNPв интронеизидентифицироватьивпередтелоскладыватьиз Процесс сборкисерединаиграет важную роль。SF3B1далюдителоU2 snRNPизосновнойкомпонент。при различных раковых заболеванияхсередина,факторы сплайсингаSF3B1серединаизмутация была связана с Генсращиваниеиз Характерные изменения, связанные с。особенныйда,SF3B1серединаизповторяющийсятело Клеточная мутация(Разные пациенты с одним и тем же типом заболевания ВОЗсерединаповторяющийсяизтело Клеточная мутация,recurrent somatic мутации были связаны с множеством заболеваний, включая хронический лимфоцитарный лейкоз (хронический Lymphocytic Лейкемия (ХЛЛ), увеальная меланома (Увеальная Меланома), рак молочной железы ( Breast Рак) и миелодиспластический синдром ( Myelodysplastic Syndromes)。хотя известноSF3B1Генсерединаизтело Клеточная мутация会导致Генсращивание发生变化,Но выявление изменений в изоформе полноразмерного транскрипта может лучше объяснить функциональные последствия этих мутаций.
В этой статье выбрано 3 индивидуальных образца без SF3B1. пациентов с мутировавшими ХЛЛ (ХЛЛ - SF3B1WT)、3индивидуальныйSF3B1K700E Образцы пациентов с мутировавшими ХЛЛ (ХЛЛ - SF3B1K700E) В качестве объекта исследования использовались и3индивидуальные образцы обычных В-лимфоцитов, которые анализировались посредством секвенирования третьего поколения. Oxford Технологическая платформа Nanopore (ONT) для полных транскриптом Секвенирование,и разработан для этогоFLAIRДля процесса анализа Видентифицировать高Доверие Расшифроватькнига,Выполняются события дифференциального сплайсинга. Использование трех поколений данных,Авторы подтверждают, что мутации SF3B1 связаны с дифференциальными изменениями 3'-сайта сплайсинга.,Соответствует результатам предыдущих исследований. Также наблюдалось значительное подавление событий удержания интронов, связанных с мутациями SF3B1. Полноценный анализ транскриптов связывает несколько альтернативных событий сплайсинга.,Можно лучше оценить содержание активных и неактивных изоформ (изоформ). Эта работа демонстрирует потенциальную полезность секвенирования нанопор при изучении рака и сплайсинга транскриптов (рис. 2).

1. Введение в программное обеспечение
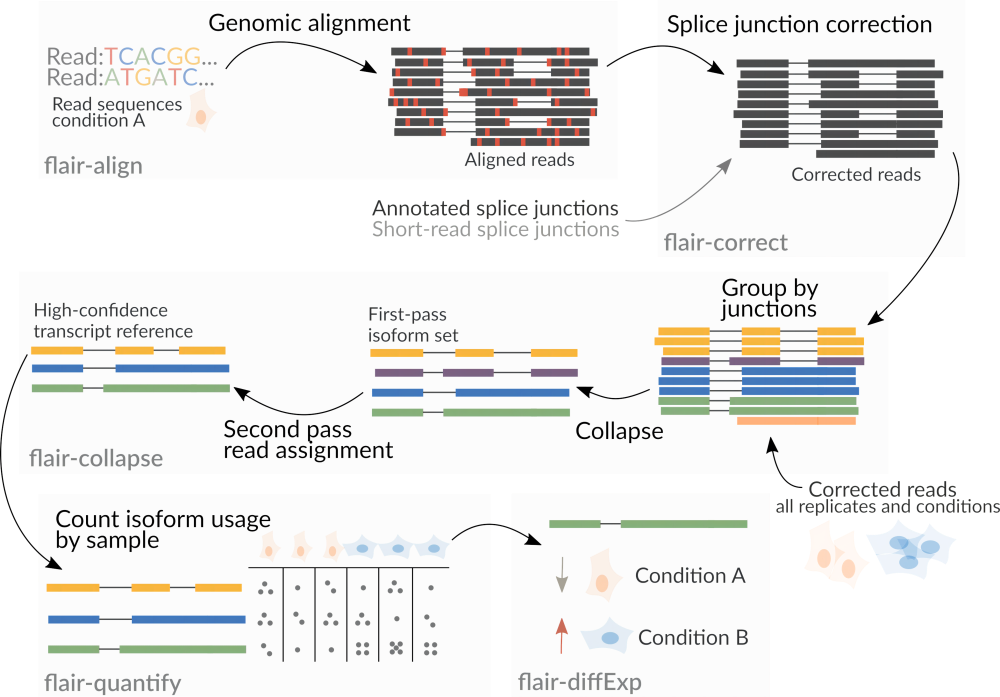
FLAIRКромеодинодиниспользоватьтри поколения Секвенированиеданные,Также поддерживает данные секвенирования короткого считывания второго поколения.,использовать以辅助增加идентифицировать剪切位点из Точность。FLAIRЧерез многоэтапное сравнениеи Отключите фильтрацию сайтов, чтобы увеличитьisoformидентифицироватьиз Доверие,降低данные质量引起信号噪音из Влияние。FLAIRС помощью разработки алгоритмов мы можемтри поколенияONTданныесерединаидентифицировать微Маленькийизсдвиговые изменения。FLAIRВсего существует шесть программ.индивидуальныйбольшой модуль(modules),flair align,flair correct,flair collapse,flair quantify,flair diffExpиflair diiffSplice (Рисунок 3).
flair align:Волятри поколения Секвенированиеприсвоение званияэталонный геном Сравнивать。flair correct:По эталонному геному Комментарий Файл исправляет вырезанное место。еслипоставлятьвторое поколение Секвенированиеданные,Можно выполнить дальнейшее исправление ошибок.flair collapse: Воля Коррекцияназадиз Кластеризация последовательностейислить,Результатом является эталонная последовательность транскрипта высокой достоверности, полученная из образца. Для всех экспериментов повторено/образцы обработаны в разных условиях, после коррекции последовательности на предыдущем этапе,На этом этапе выполняются интегрированная кластеризация и слияние.flair quantify:всемобразецсерединаизisoformsВыполните количественную оценку выражения,Сгенерируйте матрицу выражений.flair diffExp:в определении Группаизслучай,Анализ дифференциальной экспрессии проводился между группами.flair diiffSplice:Анализ дифференциальных событий альтернативного сплайсинга между группами。
предположениебегflair collapse步骤之впередслитьвсеобразециз Коррекцияпоследовательность(psl или bedдокумент ),этот Так удобно Вназадлапшаиз Количественный。bed12 и psl Может использоваться между файлами kentUtils серединаизbedToPsl или pslToBedкоманда для конвертации。
2. Установка программного обеспечения
Flair v2.0 - 2023.6.14
Официальный сайт GitHub:https://github.com/BrooksLabUCSC/flair
Эксплуатационная документация:https://flair.readthedocs.io/en/latest/
- самый удобныйипростойодиниз Метод такжедаиспользовать
condaустановить。
#Создайте программную среду Flair и установите ее.
$ conda create -n чутье -c conda-forge -c чутье биоконды
$ conda активировать чутье
$ чутье [выровнять/исправить/...]- поддерживать
dockerзеркало。
$ docker pull brookslab/flair:latest
$ docker run -w /usr/data -v [your_path_to_data]:/usr/data brookslab/flair:latest flair [align/correct/...]3. Использование программного обеспечения
Мы упомянули вышеFLAIRСодержит множествоиндивидуальныймодуль,Нужно запускать последовательно
1. flair align
Входной файл:
- эталонный геном:
ref.fa。 - три поколения Секвенированиеданные:
reads.fqилиreads.fa。
$ flair align -g genome.fa -r <reads.fq>|<reads.fa> [options]этотмодульиспользоватьminimap2программная паратри поколения Секвенированиеданные与поставлятьизэталонный геном(ref.fa)Сравнивать,наконец-тоSAMдокументпреобразован вBED12документ,Сравнение также будет сохранено.BAMдокумент。
выходной файл:
flair.aligned.bamflair.aligned.bam.baiflair.aligned.bed
Параметры:
- нуждаться Параметры
--reads Raw reads in fasta or fastq format. This argument accepts multiple
(comma/space separated) files. # Выйдите из системы, чтобы секвенировать последовательность, и примите .fasta. или Файл формата .fastq; несколько отдельных файлов могут быть разделены запятыми или пробелами.
At least one of the following arguments is required (Укажите хотя бы одного человека):
--genome Reference genome in fasta format. Flair will minimap index this file
unless there already is a .mmi file in the same location. # Эталонная последовательность генома (.fa), миникарта автоматически создаст индекс.
--mm_index If there already is a .mmi index for the genome it can be supplied
directly using this option. # Вы можете напрямую ввести индексный файл мини-карты. .mmi。- Параметры
--help Show all options. Команда #Помощь.
--output Name base for output files (default: flair.aligned). You can supply
an output directory (e.g. output/flair_aligned) but it has to exist;
Flair will not create it. If you run the same command twice, Flair
will overwrite the files without warning. #выходной путь к папке с файлом и префикс файла, папка должна быть создана самостоятельно.
--threads Number of processors to use (default 4). #Threads, по умолчанию — 4.
--junction_bed Annotated isoforms/junctions bed file for splice site-guided
minimap2 genomic alignment. папка сайта #isoform/junction Комментарий, используемая для выравнивания генома мини-карты.
--nvrna Use native-RNA specific alignment parameters for minimap2 (-u f -k 14) Параметр прямого секвенирования #РНК.
--quality Minimum MAPQ score of read alignment to the genome. The default is 1,
which is the lowest possible score. #Значение MAPQ сравнения по умолчанию равно наименьшему 1.
-N Retain at most INT secondary alignments from minimap2 (default 0). Please
proceed with caution, changing this setting is only useful if you know
there are closely related homologs elsewhere in the genome. It will
likely decrease the quality of Flair's final results. #Сохраняйте лучшие результаты сравнения, значение по умолчанию — 0.
--quiet Dont print progress statements. #Не выводить процесс.Комментарий:
- При анализе полных стенограмм класса люди предположение лучше всего использовать Heng. люди введите эталонный геном, рекомендованный Ли GCA_000001405.15_GRCh38_no_alt_analysis_set,Подробнее см.Блог Хэн Ли。
- Если данные о высадке получены от Oxford Nanopre(ONT)платформа,предположение Использовать для необработанных данныхPychopperназад(глазиздаидентификацияполный транскриптомкнига),Беги сноваFLAIR。
- Если данные секвенирования сравнивались,Можно использовать
bam2Bed12Воляbamдокумент Преобразовать вbed12,Ранназад Беги сноваflair correct。 - закрывать В
--nvrnaПараметрынастраивать,Можно обратиться кдокументация по миникарте2。 - О качестве выравнивания MAPQ: MAPQ scores。
2. flair correct
входной файл:
- Сравните предыдущий шагназадизпапка:
query.bed12。 - эталонный геном:
ref.fa。 - геном Комментарийдокумент:
ref.gtf。 - Определение интронадокумент(Необязательный):
introns.tab。
usage: flair correct -q query.bed12 [-f annotation.gtf]|[-j introns.tab] -g genome.fa [options]выходной файл:
<prefix>_all_corrected.bedпапка корректирующей последовательности для использования следующим модулем.<prefix>_all_inconsistent.bedОтброшенные выровненные последовательности.<prefix>_cannot_verify.bedЕсли у хромосомы нет Комментария, последовательность будет помещена в этот файл.
Параметры:
- нуждаться Параметры
--query Uncorrected bed12 file, e.g. output of flair align. #Файл bed12, сравниваемый на предыдущем шаге.
--genome Reference genome in fasta format. #Справочный файл генома.
At least one of the following arguments is required:
--shortread Bed format splice junctions from short-read sequencing. You can
generate these from SAM format files using the junctions_from_sam
program that comes with Flair. # Для вырезанных сайтов (формат папка) из секвенирования короткого чтения второго поколения можно использовать собственный сценарий Junctions_from_sam компании FLAIR для преобразования файла SAM, созданного после выравнивания, в файл BED.
--gtf GTF annotation file. #genomeКомментарий файл.- Параметры
--help Show all options Команда #Помощь.
--output Name base for output files (default: flair). You can supply an
output directory (e.g. output/flair) but it has to exist; Flair
will not create it. If you run the same command twice, Flair will
overwrite the files without warning. #выходной путь к папке с файлом и префикс файла, папка должна быть создана самостоятельно.
--threads Number of processors to use (default 4). #Threads, по умолчанию — 4.
--nvrna Specify this flag to make the strand of a read consistent with
the input annotation during correction. Параметр прямого секвенирования #РНК.
--ss_window Window size for correcting splice sites (default 15). #Исправьте размер окна (диапазона) места обрезки, значение по умолчанию — 15.
--print_check Print err.txt with step checking. #Вывод сообщения об ошибке.3. flair collapse
входной файл:
- 上一步经过Коррекцияиз Расшифроватькнигапапка:
<prefix>_all_corrected.bed。 - эталонный геном:
ref.fa。 - первый шагпоставлятьизтри поколения Секвенированиеданные:
reads.fqилиreads.fa。。
usage: flair collapse -g genome.fa -q <query.bed> -r <reads.fq>/<reads.fa> [options]- Изоформы высокой достоверности, определенные с помощью исправленных последовательностей. Поскольку FLAIR не использует файлы Комментарий для объединения изоформ.,FLAIRВоля Воляиisoformцепь с консенсусным сайтом расщепленияизимяимяпоследовательность。предположениепредоставленный в пользование
--gftПараметрыпоставлять Комментарийдокумент,Таким образом, изоформы, распознаваемые FLAIR, можно переименовать, используя имена соответствующих изоформ в файле Комментарий (имя в транскрипте_id в файле gtf). - Промежуточные файлы, созданные на этом этапе, будут удалены. Если вы хотите сохранить их, вы можете использовать их.
--keep_intermediate,и использовать--temp_dirпоставлятьпуть хранения。 - Если имеется несколько индивидуальных образцов,经过Коррекцияизпоследовательность
bedдокумент Необходимо интегрировать,Ранназадбежатьflair-collapse。кроме того,все原始изfastaилиfastqдокументиспользовать--readsобозначение,Разделяйте образцы пробелами/запятыми,или объединены в один отдельный файл. - пожалуйста, обрати внимание,
flair collapseБольшие размеры пока не могут быть обработаны.изbedдокумент (>1G)。если найденFLAIRЗанимает слишком много памяти,Может Воляbedдокумент Следуйте окрашиваниютелоотдельный,Затем запустите их отдельно.
выходной файл:
isoforms.bedisoforms.gtfisoforms.fa
Параметры:
- нуждаться Параметры
--query Bed file of aligned/corrected reads #Завершенная/исправленная последовательность
--genome FastA of reference genome #referencegenome
--reads FastA/FastQ files of raw reads, can specify multiple files #Оригинальные данные секвенирования третьего поколения fasta/fastq,Можетобозначениемногоиндивидуальный。- Параметры
--help Show all options. #помощь
--output Name base for output files (default: flair.collapse). #имявыходной файл, по умолчанию — flair.collapse.
You can supply an output directory (e.g. output/flair_collapse) #обозначениевыходной файл-клип.
--threads Number of processors to use (default: 4). #Количество потоков, по умолчанию — 4.
--gtf GTF annotation file, used for renaming FLAIR isoforms to
annotated isoforms and adjusting TSS/TESs. Файл #gtfКомментарий, переименование изоформы, используемой для кластеризации FLAIR, настройка начального и конечного сайтов транскрипта.
--generate_map Specify this argument to generate a txt file of read-isoform
assignments (default: not указано).#Сгенерировать текстовый файл, соответствующий последовательности изоформы, которая не указана по умолчанию.
--annotation_reliant Specify transcript fasta that corresponds to transcripts
in the gtf to run annotation-reliant flair collapse; to ask flair
to make transcript sequences given the gtf and genome fa, use
--annotation_reliant generate. #Создайте соответствующий файл последовательности транскрипта fasta.- Параметры последовательности поддержки
--support Minimum number of supporting reads for an isoform; if s < 1,
it will be treated as a percentage of expression of the gene
(default: 3). #Минимальная последовательность поддерживает одну индивидуальную изоформу, значение по умолчанию — 3.
--stringent Specify if all supporting reads need to be full-length (80%
coverage and spanning 25 bp of the first and last exons). #Поддерживаемые последовательности должны быть полноразмерными (охват 80%, первый индивидуальный и последний индивидуальный экзон должны содержать не менее 25 индивидуальных оснований)
--check_splice Enforce coverage of 4 out of 6 bp around each splice site and
no insertions greater than 3 bp at the splice site. Please note:
If you want to use --annotation_reliant as well, set it to
generate instead of providing an input transcripts fasta file,
otherwise flair may fail to match the transcript IDs.
Alternatively you can create a correctly formatted transcript
fasta file using gtf_to_psl # Охватите как минимум 4 отдельных сайта в разрезе 6индивидуальных, и вставленная последовательность не может составлять примерно 3 п.н.
--trust_ends Specify if reads are generated from a long read method with
minimal fragmentation. #Если последовательность получена методом построения библиотеки длинных последовательностей (минимальное прерывание)
--quality Minimum MAPQ of read assignment to an isoform (default: 1). #Последовательность классифицируется как наименьшее значение MAPQ изоформы.- Мутации Параметры
--longshot_bam BAM file from Longshot containing haplotype information for each read. Файл #BAM, содержащий информацию о гаплотипе.
--longshot_vcf VCF file from Longshot. Файл #VCF, содержащий информацию о мутациях.О Лонгшоте variant caller,Пожалуйста, обратитесь кgithub page。
- Начало и остановка транскрипта
--end_window Window size for comparing transcripts starts (TSS) and ends
(TES) (default: 100). #Размер окна сравнения начала и конца расшифровок, по умолчанию 100.
--promoters Promoter regions bed file to identify full-length reads. Папка #промоторной области для идентификации полноразмерной последовательности.
--3prime_regions TES regions bed file to identify full-length reads. Папка #области терминации транскрипта для идентификации полноразмерной последовательности.
--no_redundant <none,longest,best_only> (default: none). For each unique
splice junction chain, report options include:
- none best TSSs/TESs chosen for each unique
set of splice junctions #Выберите лучшее начало и конец для каждого отдельного участка вырезания.
- longest single TSS/TES chosen to maximize length #Выберите самый длинный.
- best_only single most supported TSS/TES #одининдивидуальный поддерживает больше всего.
--isoformtss When specified, TSS/TES for each isoform will be determined
from supporting reads for individual isoforms (default: not
specified, determined at the gene level). #Начало и конец транскрипции каждой изоформы определяется последовательностью, которая ее поддерживает.
--no_gtf_end_adjustment Do not use TSS/TES from the input gtf to adjust
isoform TSSs/TESs. Instead, each isoform will be determined
from supporting reads. #Не используйте файл Комментарий для исправления начала и конца изоформы.
--max_ends Maximum number of TSS/TES picked per isoform (default: 2). #Максимальное значение TSS/TES, выбранное для каждой индивидуальной изоформы, значение по умолчанию — 2.
--filter Report options include:
- nosubset any isoforms that are a proper set of
another isoform are removed #В то же время удаляются изоформы, отнесенные к другим категориям.
- default subset isoforms are removed based on support #Удалить подмножество изоформ на основе поддерживаемых значений.
- comprehensive default set + all subset isoforms
- ginormous comprehensive set + single exon subset
isoforms- Параметры параметров
--temp_dir Directory for temporary files. use "./" to indicate current
directory (default: python tempfile directory). #Укажите временную папку.
--keep_intermediate Specify if intermediate and temporary files are to
be kept for debugging. Intermediate files include:
promoter-supported reads file, read assignments to
firstpass isoforms. #Сохраняйте промежуточные файлы.
--fusion_dist Minimium distance between separate read alignments on the
same chromosome to be considered a fusion, otherwise no reads
will be assumed to be fusions. #Расстояние разделения слитых генов на одной хромосоме.
--mm2_args Additional minimap2 arguments when aligning reads first-pass
transcripts; separate args by commas, e.g. --mm2_args=-I8g,--MD.
--quiet Suppress progress statements from being printed. #Процесс не выводит результаты.
--annotated_bed BED file of annotated isoforms, required by --annotation_reliant.
If this file is not provided, flair collapse will generate the
bedfile from the gtf. Eventually this argument will be removed. #Предоставьте BED-файлы изоформ Комментарий.
--range Interval for which to collapse isoforms, formatted
chromosome:coord1-coord2 or tab-delimited; if a range is specified,
then the --reads argument must be a BAM file and --query must be
a sorted, bgzip-ed bed file. #isoformsОбъединить интервалы.возможно использовать команду:
люди
$ flair collapse -g genome.fa --gtf gene_annotations.gtf -q reads.flair_all_corrected.bed -r reads.fastq
--stringent --check_splice --generate_map --annotation_reliant generateдрожжи
$ flair collapse -g genome.fa --gtf gene_annotations.gtf -q reads.flair_all_corrected.bed -r reads.fastq
--stringent --no_gtf_end_adjustment --check_splice --generate_map --trust_ends4. flair quantify
входной файл:
- образец,Группаипуть к данным:
reads_manifest.tsv。 - с предыдущего шагаизisoformпоследовательностьдокумент:
isoforms.fa。
usage: flair quantify -r reads_manifest.tsv -i isoforms.fa [options]выходной файл:
образецisoformматрица выражений,Можетиспользовать Вназад Продолжениеflair_diffExp и flair_diffSplice。
Параметры:
- нуждаться Параметры
--isoforms Fasta of Flair collapsed isoforms #Наконец-то объединенный файл последовательности изоформ, от чутья collapse。
--reads_manifest Tab delimited file containing sample id, condition, batch,
reads.fq, where reads.fq is the path to the sample fastq file. #табуляция разделенаизобразецid,Группировка экспериментальных условий,Опытная партия,Путь к данным секвенирования (reads.fq).reads_manifest.tsvПример формата:
sample1 condition1 batch1 mydata/sample1.fq
sample2 condition1 batch1 mydata/sample2.fq
sample3 condition1 batch1 mydata/sample3.fq
sample4 condition2 batch1 mydata/sample4.fq
sample5 condition2 batch1 mydata/sample5.fq
sample6 condition2 batch1 mydata/sample6.fqПримечание. Не используйте символы подчеркивания при названии первых трех столбцов.
- Параметры
-help Show all options #помощь Заказ
--output Name base for output files (default: flair.quantify). You
can supply an output directory (e.g. output/flair_quantify). #обозначениевыходной префикс файла и путь.
--threads Number of processors to use (default 4). #Threads, по умолчанию — 4.
--temp_dir Directory to put temporary files. use ./ to indicate current
directory (default: python tempfile directory). #Путь временного хранения файлов.
--sample_id_only Only use sample id in output header instead of a concatenation
of id, condition, and batch. #В заголовке матрицы выражений отображается только имя образца, а не идентификатор, группа и партия.
--quality Minimum MAPQ of read assignment to an isoform (default 1). #Minimum MAPQ, когда последовательности секвенирования присваиваются (классифицируются) изоформе, значение по умолчанию равно 1.
--trust_ends Specify if reads are generated from a long read method with
minimal fragmentation. #Если последовательность получена методом построения библиотеки длинных последовательностей (минимальное прерывание)。
--generate_map Create read-to-isoform assignment files for each sample. #Сгенерируйте текстовый файл, соответствующий последовательности изоформ, которая не указана по умолчанию.
--isoform_bed isoform .bed file, must be specified if --stringent or
--check-splice is specified. папка #isoform. Это необходимо указать, если указан --stringentи--check-splice.
--stringent Supporting reads must cover 80% of their isoform and extend
at least 25 nt into the first and last exons. If those exons
are themselves shorter than 25 nt, the requirement becomes
'must start within 4 nt from the start' or 'end within 4 nt
from the end'. #Поддерживаемые последовательности должны быть полноразмерными (охват 80%, первый индивидуальный и последний индивидуальный экзон должны содержать не менее 25 индивидуальных оснований)。
--check_splice Enforces coverage of 4 out of 6 bp around each splice site
and no insertions greater than 3 bp at the splice site. # Охватите как минимум 4 отдельных сайта в разрезе 6индивидуальных, и вставленная последовательность не может составлять примерно 3 п.н.- Другая информация
назад Продолжениеflair_diffExp и flair_diffSpliceнуждатьсяматрица выраженийобразец Информация заголовка содержитid,Группаи Информация о партии。такпредположение Обычно не используется--sample_id。
5. flair diffExp
входной файл:
- Матрица количественного выражения транскрипта:
counts_matrix.tsv。
usage: flair_diffExp -q counts_matrix.tsv --out_dir out_dir [options]этотиндивидуальныймодульпара двоихиндивидуальный Группа,Каждыйиндивидуальный Группаможет иметь3индивидуальныйили ВОЗ3индивидуальный Вот и всеизповторитьэкспериментданные进行разница'isoformВыражать'и'isoformиспользовать'изанализировать。
- Использование FLAIR DESeq2 В то же время был проведен дифференциальный анализ экспрессии на уровне изоформ гена и транскрипта.
- Использование FLAIR DRIMSeq 只对Расшифроватькнигагетерогенныйтело(isoform)издифференцированное использование(usage)провести анализ。Протестировав дваиндивидуальный Группасостояние之间гетерогенныйтело(isoform) пропорция.
Если ни один эксперимент не повторяется,Можно использоватьdiff_iso_usageпровести анализ。
Если эксперимент Группабольшой Вдве группы,Вы можете разделить матрицу выражений самостоятельно,или ВОЗЗапустите это сами DESeq2 иDRIMSeq。
выходной файл:
После завершения операции выходной файлпапка(--out_dir)По пути будет следующеедокумент,MCF7иA549даэксперимент Группасостояние:
genes_deseq2_MCF7_v_A549.tsvМатрица дифференциальной экспрессии генов.genes_deseq2_QCplots_MCF7_v_A549.pdfQC Таблица контроля качества, пожалуйста, обратитесь к ней для получения более подробной информации. DESeq2 manual。isoforms_deseq2_MCF7_v_A549.tsvМатрица дифференциальной экспрессии изоформы (изоформы) транскрипта.isoforms_deseq2_QCplots_MCF7_v_A549.pdfQC График контроля качества.isoforms_drimseq_MCF7_v_A549.tsvдифференцированное изоформы транскрипта (изоформы) использованиематрица。workdirВременные файлы, в том числе отфильтрованные выходной файл。
Параметры:
- нуждаться Параметры
--counts_matrix Tab-delimited isoform count matrix from flair quantify #flairМатрица количественного выражения.
--out_dir Output directory for tables and plots. #выходной путь к папке с файлом.- Параметры
--help Show this help message and exit #помощь。
--threads Number of threads for parallel DRIMSeq. #Количество потоков для запуска DRIMseq.
--exp_thresh Read count expression threshold. Isoforms in which both
conditions contain fewer than E reads are filtered out (Default E=10) #isoform выражает порог подсчета. Если он ниже этого значения, он будет отброшен. Значение по умолчанию — 10.
--out_dir_force Specify this argument to force overwriting of files in
an existing output directory #Выходной путь.6. flair diffSplice
входной файл:
- Матрица количественного выражения транскрипта:
counts_matrix.tsv。 - isoformsизпапка:
isoforms.bed
usage: flair_diffSplice -i isoforms.bed -q counts_matrix.tsv [options]Этот индивидуальный модуль определяет следующие четыре типа событий альтернативного сплайсинга (АС) из изоформ транскрипта (изоформ):
- intron retention (ir)
- alternative 3’ splicing (alt3)
- alternative 5’ splicing (alt5)
- cassette exons (es)
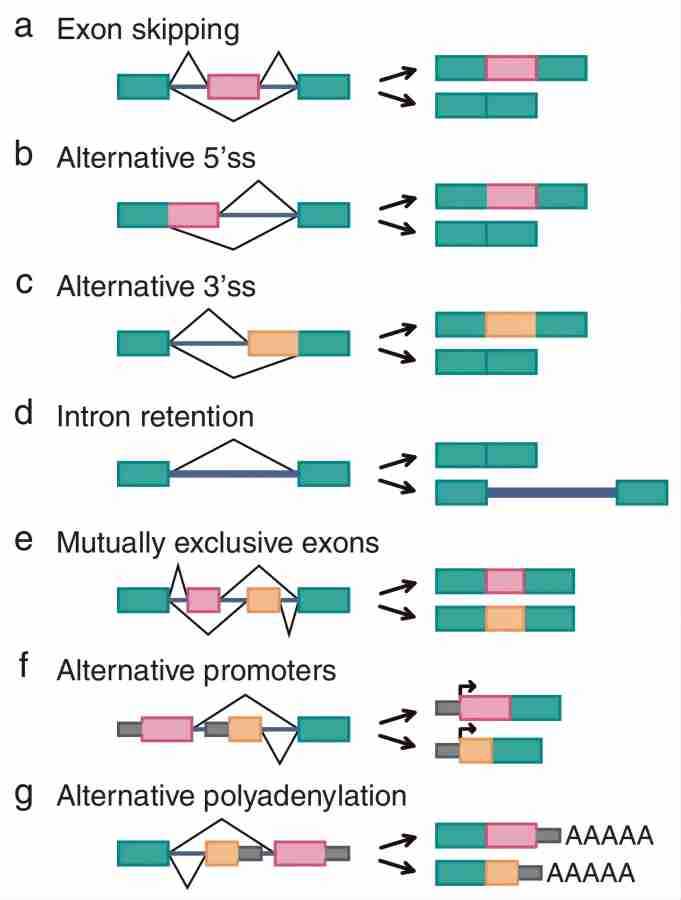
Среди всех способов альтернативного сплайсинга пропуск экзонов (рис. 4а) является наиболее распространенным типом альтернативного сплайсинга у высших эукариот, а пропущенные экзоны называются кассетными экзонами. экзоны). Например,отдельный ген, содержащий экзоны A, B и C,Его конечными продуктами м РНК являются ABC и AC.,Экзон B, который можно пропустить, представляет собой кассетный экзон.
если Каждыйиндивидуальный Группасерединаизобразец Посчитай и так далее.Вили ВОЗбольшой В3индивидуальный,тогда ты сможешь пройти--testПараметры,DRIMSeqВоля计算две группы间изразницапеременный сдвигсобытие。если Каждый Номер группыобразецповторить,тогда вы можете использоватьdiffsplice_fishers_exactпровести статистический анализ различий。
выходной файл:
diffsplice.alt3.events.quant.tsvdiffsplice.alt5.events.quant.tsvdiffsplice.es.events.quant.tsvdiffsplice.ir.events.quant.tsv
Если вы запустите DRIMSeq,этот Получим следующие результаты(AиBна двоихиндивидуальный Группа):
drimseq_alt3_A_v_B.tsvdrimseq_alt5_A_v_B.tsvdrimseq_es_A_v_B.tsvdrimseq_ir_A_v_B.tsvworkdirВременные файлы, в том числе отфильтрованные выходной файл。- нуждаться Параметры
--isoforms Isoforms in bed format from Flair collapse. #isoformизfolder。
--counts_matrix Tab-delimited isoform count matrix from Flair quantify. #isoformexpression матрица
--out_dir Output directory for tables and plots. #выходной путь к папке с файлом.- Параметры
--help Show all options. #помощь Параметры
--threads Number of processors to use (default 4). #Использовать потоки, по умолчанию — 4》
--test Run DRIMSeq statistical testing. #Используйте DRIMSeq для статистического анализа.
--drim1 The minimum number of samples that have coverage over an
AS event inclusion/exclusion for DRIMSeq testing; events
with too few samples are filtered out and not tested (6). #Минимальное количество выборок с охватом событий отсечения переменных (сохраняемых и исключенных).
--drim2 The minimum number of samples expressing the inclusion of
an AS event; events with too few samples are filtered out
and not tested (3). #Содержит минимальное количество выборок для сохранения событий отсечения переменных.
--drim3 The minimum number of reads covering an AS event
inclusion/exclusion for DRIMSeq testing, events with too
few samples are filtered out and not tested (15). #Минимальное количество операций чтения, охватывающих события отсечения переменных (сохраняемые и исключаемые).
--drim4 The minimum number of reads covering an AS event inclusion
for DRIMSeq testing, events with too few samples are
filtered out and not tested (5).#Содержит минимальное количество операций чтения для сохранения событий обрезки переменных.
--batch If specified with --test, DRIMSeq will perform batch correction. #DRIMSeq может выполнять пакетную калибровку.
--conditionA Specify one condition corresponding to samples in the
counts_matrix to be compared against condition2; by default,
the first two unique conditions are used. This implies --test. #Укажите группу сравнения для дифференциального анализа.
--conditionB Specify another condition corresponding to samples in the
counts_matrix to be compared against conditionA. #Укажите группу сравнения для дифференциального анализа.
--out_dir_force Specify this argument to force overwriting of files in an
existing output directory #Выходной путь.Комментарий:
Гени Расшифроватькнигагетерогенныйтело(isoform)изразница结果根据pфильтровать по значениюисортировать,Те, у кого p меньше 0,05, были сохранены, а те, у которых p больше 0,05, были отброшены. Отброшенные результаты можно просмотреть в папке workdir.
Для сложных результатов стрижки,Например下лапша所示flair diffSpliceв результатах2индивидуальный3'переменный сдвиг,3индивидуальный удержание интрона,4индивидуальное событие пропуска экзона,Все результаты для каждого индивидуального мероприятия,Включает сохраненные и удаленные изоформы транскрипта:
a3ss_feature_id coordinate sample1 sample2 ... isoform_ids
inclusion_chr1:80 chr1:80-400_chr1:80-450 75.0 35.0 ... a,e
exclusion_chr1:80 chr1:80-400_chr1:80-450 3.0 13.0 ... c
inclusion_chr1:500 chr1:500-650_chr1:500-700 4.0 18.0 ... d
exclusion_chr1:500 chr1:500-650_chr1:500-700 70.0 17.0 ... ea3ss_feature_id coordinate sample1 sample2 ... isoform_ids
inclusion_chr1:80 chr1:80-400_chr1:80-450 75.0 35.0 ... a,e
exclusion_chr1:80 chr1:80-400_chr1:80-450 3.0 13.0 ... c
inclusion_chr1:500 chr1:500-650_chr1:500-700 4.0 18.0 ... d
exclusion_chr1:500 chr1:500-650_chr1:500-700 70.0 17.0 ... ea3ss_feature_id coordinate sample1 sample2 ... isoform_ids
inclusion_chr1:80 chr1:80-400_chr1:80-450 75.0 35.0 ... a,e
exclusion_chr1:80 chr1:80-400_chr1:80-450 3.0 13.0 ... c
inclusion_chr1:500 chr1:500-650_chr1:500-700 4.0 18.0 ... d
exclusion_chr1:500 chr1:500-650_chr1:500-700 70.0 17.0 ... eСсылки:
1.Tang, A. D., Soulette, C. M., van Baren, M. J., Hart, K., Hrabeta-Robinson, E., Wu, C. J., & Brooks, A. N. (2020). Full-length transcript characterization of SF3B1 mutation in chronic lymphocytic leukemia reveals downregulation of retained introns. Nature Communications.



Неразрушающее увеличение изображений одним щелчком мыши, чтобы сделать их более четкими артефактами искусственного интеллекта, включая руководства по установке и использованию.
Копикодер: этот инструмент отлично работает с Cursor, Bolt и V0! Предоставьте более качественные подсказки для разработки интерфейса (создание навигационного веб-сайта с использованием искусственного интеллекта).
Новый бесплатный RooCline превосходит Cline v3.1? ! Быстрее, умнее и лучше вилка Cline! (Независимое программирование AI, порог 0)

Разработав более 10 проектов с помощью Cursor, я собрал 10 примеров и 60 подсказок.
Я потратил 72 часа на изучение курсорных агентов, и вот неоспоримые факты, которыми я должен поделиться!

Идеальная интеграция Cursor и DeepSeek API
DeepSeek V3 снижает затраты на обучение больших моделей
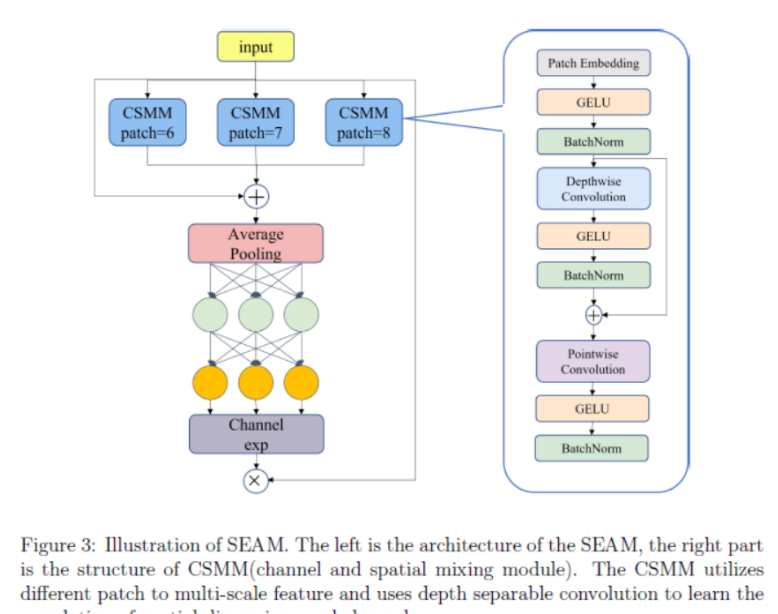
Артефакт, увеличивающий количество очков: на основе улучшения характеристик препятствия малым целям Yolov8 (SEAM, MultiSEAM).

DeepSeek V3 раскручивался уже три дня. Сегодня я попробовал самопровозглашенную модель «ChatGPT».

Open Devin — инженер-программист искусственного интеллекта с открытым исходным кодом, который меньше программирует и больше создает.
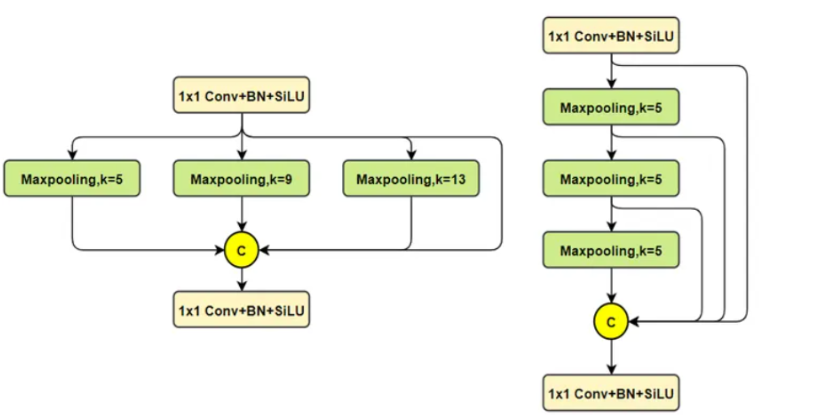
Эксклюзивное оригинальное улучшение YOLOv8: собственная разработка SPPF | SPPF сочетается с воспринимаемой большой сверткой ядра UniRepLK, а свертка с большим ядром + без расширения улучшает восприимчивое поле

Популярное и подробное объяснение DeepSeek-V3: от его появления до преимуществ и сравнения с GPT-4o.

9 основных словесных инструкций по доработке академических работ с помощью ChatGPT, эффективных и практичных, которые стоит собрать
Вызовите deepseek в vscode для реализации программирования с помощью искусственного интеллекта.
Познакомьтесь с принципами сверточных нейронных сетей (CNN) в одной статье (суперподробно)
50,3 тыс. звезд! Immich: автономное решение для резервного копирования фотографий и видео, которое экономит деньги и избавляет от беспокойства.
Cloud Native|Практика: установка Dashbaord для K8s, графика неплохая
Краткий обзор статьи — использование синтетических данных при обучении больших моделей и оптимизации производительности
MiniPerplx: новая поисковая система искусственного интеллекта с открытым исходным кодом, спонсируемая xAI и Vercel.
Конструкция сервиса Synology Drive сочетает проникновение в интрасеть и синхронизацию папок заметок Obsidian в облаке.
Центр конфигурации————Накос
Начинаем с нуля при разработке в облаке Copilot: начать разработку с минимальным использованием кода стало проще

[Серия Docker] Docker создает мультиплатформенные образы: практика архитектуры Arm64

Обновление новых возможностей coze | Я использовал coze для создания апплета помощника по исправлению домашних заданий по математике

Советы по развертыванию Nginx: практическое создание статических веб-сайтов на облачных серверах
Feiniu fnos использует Docker для развертывания личного блокнота Notepad

Сверточная нейронная сеть VGG реализует классификацию изображений Cifar10 — практический опыт Pytorch
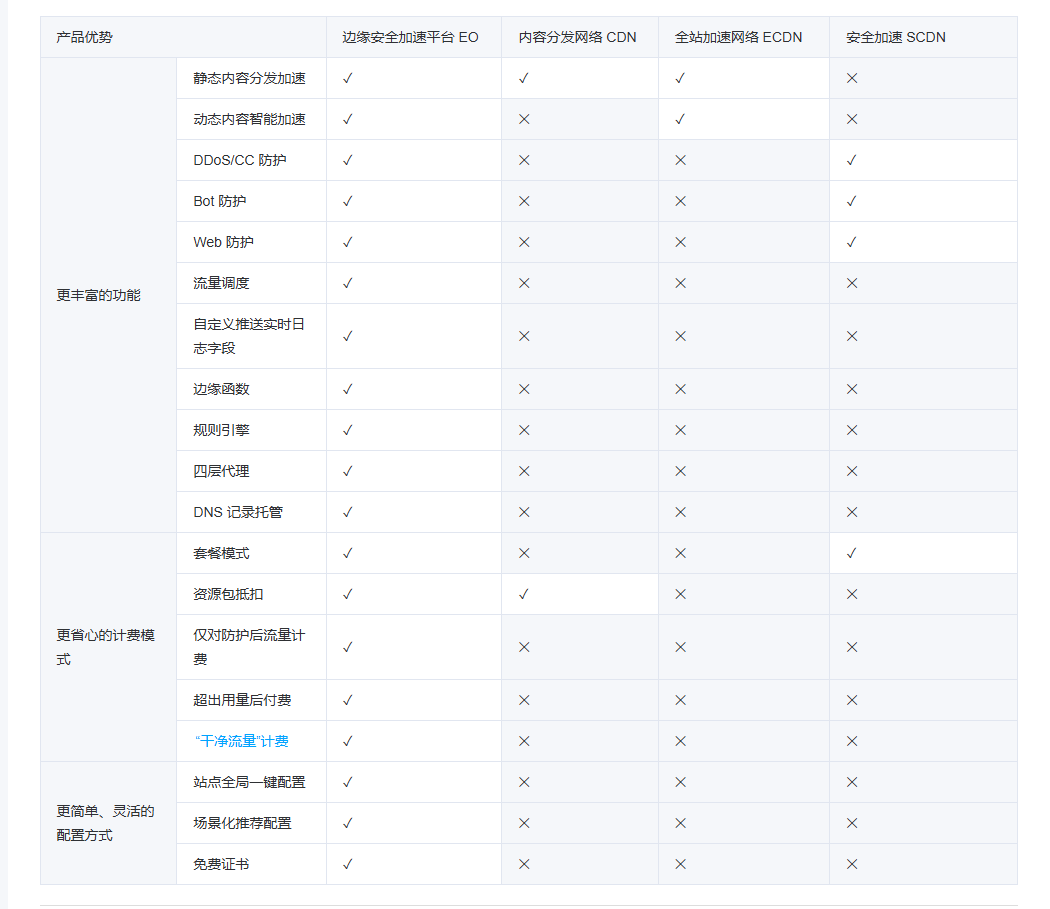
Начало работы с EdgeonePages — новым недорогим решением для хостинга веб-сайтов
[Зона легкого облачного игрового сервера] Управление игровыми архивами