полный транскриптом | три поколенияполный транскриптоманалитический процесс (PacBio & ONT )-- Bambu
Сегодня мы продолжаем представлять инструмент для аннотирования и количественной оценки транскриптов с использованием трех поколений полноразмерных данных транскриптов. - Bambu。от Сингапурского агентства по науке, технологиям и исследованиям (A-STAR) изJonathan Göke(картина1)продолжительность разработкиRNA-seqИнструменты транскриптомного анализаBambu,Опубликовано в журнале «Nature Methods» 12 июня 2023 г.,НазваниеContext-aware transcript quantification from long-read RNA-seq data with Bambu。Этот инструмент основан на машинном обучении для идентификации и характеристики новых транскриптов.,Это позволяет проводить анализ адаптивности различных видов и образцов.

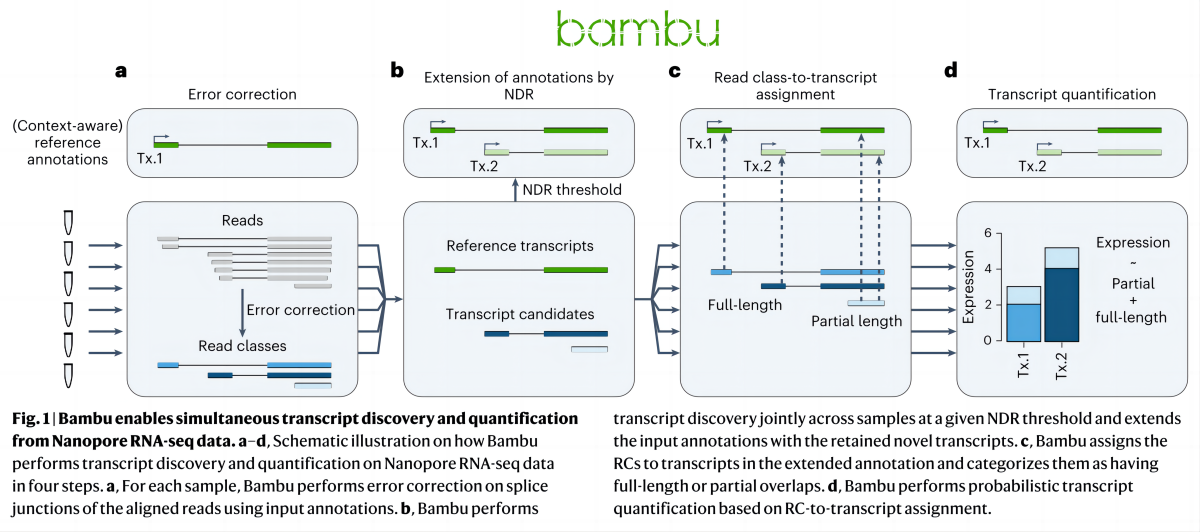
Однако большинство методов количественной оценки транскриптов основаны на файлах аннотаций фиксированных ссылок на гены;,Настоящие транскриптомы динамичны.,Надо судить по ситуации на данный момент.,Файлы статических аннотаций транскриптома содержат неактивные транскрипты (изоформы) некоторых генов.,иверно Некоторые гены имеют неполную аннотацию.。Bambu,Метод, использующий данные секвенирования длинной РНК.,Узнать Метод идентификации транскриптов на основе машинного обучения,Осуществите количественную оценку транскриптов в реальных данных секвенирования. Для выявления новых транскриптов,Bambu Предполагаемая скорость открытия новых discovery rate),объяснимый、Откалиброванный по точности отдельный параметр заменяет произвольный порог для одной выборки。Bambu Сохраняет полноразмерные и уникальные последовательности транскриптов в присутствии неактивных транскриптов (изоформ). может выполнить точную количественную оценку. По сравнению с существующими методами идентификации транскриптов,Bambu Повышенная точность достигается без потери чувствительности.。Динамическая аннотация, основанная на реальных данных, улучшает количественную оценку новых и известных транскриптов.。Воспользуйтесь преимуществом длительного чтенияRNAСеквенирование данных и машинное обучение,Bambu Облегчает точную идентификацию и количественную оценку транскриптов (рис. 2).

1. Введение в программное обеспечение
Bambu это Воспользуйтесь преимуществом длительного чтенияRNA-SeqДанные для идентификации и количественной оценки транскриптов в нескольких образцахпакет R。Выполнение сравнения последовательностейверноназад,Можно использоватьBambu Получите известные и новые транскрипты и уровни экспрессии генов.。Bambu Выходные данные можно использовать непосредственно для визуализации и последующего анализа, например, дифференциальной экспрессии генов или использования транскриптов.
Bambu Процесс анализа можно разделить на четыре этапа (рис. 3. a-d):первый,Использование вероятностной модели,Используйте справочные аннотации、Геномные последовательности и особенности, извлеченные из данных (подробные характеристики можно найти в разделе «Методы») для коррекции соотношения областей или сайтов соединения РНК. Исправленные последовательности, использующие один и тот же сайт разреза, были объединены в одну последовательность. (Read class, RCs)。Шаг 2,Интегрируйте объединенные последовательности из всех образцов (Читать классы), а также новый уровень открытий в выборках открытия, Чтение меньше). определенные порога. Последовательности класса NDR классифицируются как новые транскрипты.,и добавьте новую аннотированную расшифровку к аннотации эталонной расшифровки,Сформируйте библиотеку аннотаций на основе реальных образцов.(Расширенные оригинальные справочные примечания)。Шаг 3,Аннотация гена с использованием расширенной ссылки,Переклассифицируйте каждую последовательность классов Read и дайте ей название. в этом процессе,С тех порверно Неточные совпадения последовательностей, вызванные ошибками, можно исправить.。Шаг 4,Аннотация гена с использованием расширенной ссылки,верно Транскрипты в каждом образце для окончательного количественного определения,Получите матрицу экспрессии генов/транскриптов.
2. Установка программного обеспечения
Bambu: https://github.com/GoekeLab/bambu
Версия: v3.2.4
1. Установите Bambu с помощью Bioconductor.
существоватьRсередина,илиRstudio,илиRstudio-serverсредасередина Запустите следующую команду для установки,Установка завершена, как показано на рисунке 4.
if (!requireNamespace("BiocManager", quietly = TRUE)) install.packages("BiocManager") #Если BiocManager уже установлен, вы можете игнорировать эту строку команды
BiocManager::install("bambu")
предположение:потому чтополный транскриптом Объем данных обычно велик, а объем вычислений велик,Локальное программное обеспечение R, развернутое на обычном настольном компьютере или на вашем собственном ноутбуке.,Может занимать много памяти и вычислительных ресурсов.。предложение использовать сервер,развертывать Rstudio-server,Это подойдетсуществовать Хранение данных на сервереи Рассчитано。Специальная установкаразвертывать Пожалуйста, обратитесь к официальному сайту за инструкциями.:https://posit.co/download/rstudio-server/。
2. Проверьте установку Bambu.
Используйте встроенные тестовые данные, чтобы подтвердить успешную установку Bambu.
#Загрузка пакета бамбука
library(bambu)
#Выполните следующие команды, чтобы импортировать данные секвенирования, эталонный геном, файлы аннотаций эталонного генома и выполнить полный транскриптоманализировать
test.bam <- system.file("extdata", "SGNex_A549_directRNA_replicate5_run1_chr9_1_1000000.bam", package = "bambu") #Читаем данные секвенирования
fa.file <- system.file("extdata", "Homo_sapiens.GRCh38.dna_sm.primary_assembly_chr9_1_1000000.fa", package = "bambu") #Читаем эталонный геном
gtf.file <- system.file("extdata", "Homo_sapiens.GRCh38.91_chr9_1_1000000.gtf", package = "bambu") #Читаем эталонный файл аннотации гена
bambuAnnotations <- prepareAnnotations(gtf.file) #Предварительная обработка аннотаций эталонного генома
se <- bambu(reads = test.bam, annotations = bambuAnnotations,genome = fa.file) #полный транскриптоманализироватьПосле запуска последней строки мы получаем --- Start isoform quantification --- и--- Finished running Bambu ---,Это означает, что Bambu запущен и успешно установлен.
3. Использование программного обеспечения
Входной файл в режиме по умолчанию:
- пройтииэталонный геном Сравниватьвернофайл последовательности,
.bamФормат файла。 - эталонный файл аннотации гена,
.gtfдокумент。 - эталонный геномдокумент,
.faдокумент。
1. Подготовьте файлы последовательностей эталонного генома.
Потому что документация Bambu не предлагает лучшего метода, чем верный.,Поэтому здесь используется обычно используемая длина.(long-read)Сравниватьвернопрограммное обеспечениеminimap2。
#Выберите соответствующую команду запуска на основе платформы секвенирования третьего поколения и метода построения библиотеки, одноэтапного метода.
$ minimap2 -ax splice:hq -uf ref.fa iso-seq.fq | samtools sort -@ 12 -o align.bam --write-index - # PacBio Iso-seq/traditional cDNA
$ minimap2 -ax splice ref.fa nanopore-cdna.fa | samtools sort -@ 12 -o align.bam --write-index - # Nanopore 2D cDNA-seq
$ minimap2 -ax splice -uf -k14 ref.fa direct-rna.fq | samtools sort -@ 12 -o align.bam --write-index - # Nanopore Direct RNA-seq
#Пошаговый пример
$ minimap2 -ax splice:hq -uf ref.fa iso-seq.fq | samtools view -@ 12 -bS | samtools sort -@ 12 -o align.bam # PacBio Iso-seq/traditional cDNA
$ minimap2 -ax splice ref.fa nanopore-cdna.fa | samtools view -@ 12 -bS | samtools sort -@ 12 -o align.bam # Nanopore 2D cDNA-seq
$ minimap2 -ax splice -uf -k14 ref.fa direct-rna.fq | samtools view -@ 12 -bS | samtools sort -@ 12 -o align.bam # Nanopore Direct RNA-seq
#вернобам Индексация файлов
$ samtools index align.bamУведомление:
- PacBioплатформаиONTДанные, генерируемые платформой,Конкретные параметры различаются,Подробности см. в документации по использованию minimap2.
- Используемый здесь эталонный геном должен быть унифицирован с геномом, использованным Бамбу.
- Нужно заранее установить
samtools,Обратите внимание на установленную версию. - потому чтоminimap2Результат:
.samФормат файла,Так что используйтеsamtoolsВоля.samпреобразован в.bam,и использоватьsamtools sortверно.bamСортировать,Ранназадиспользоватьsamtools indexиндекс。Выше показан поворот в один шаг.bamи Передача шаг за шагом.bamПример。 samtools v1.18Версия--write-index -параметры;samtools v1.9Нет версии。
2. Прочтите файл .bam образца последовательности.
#Может читать несколько файлов примеров одновременно
samples <- c(S1.bam, S1.bam, S1.bam)3. Подготовьте файлы эталонного генома.
Если людии Мыши могутсуществоватьGencodeскачать,Если вы встретите другие виды, вы можете обратиться кEnsemblскачать。
4. Подготовка аннотации эталонного генома.
annotations <- prepareAnnotations( *.gft )
#Сохраняем файл комментариев как файл rds
saveRDS(annotations, ”/path/to/annotations.rds” )
#Прочитайте файл комментариев в следующий раз
annotations <- readRDS("/path/to/annotations.rds")5. Беги Бамбу
se <- bambu(reads = samples, annotations = annotations, genome = genome.fa)6. Выявляйте только новые транскрипты, а не оценивайте их количественно
se.discoveryOnly <- bambu(reads = samples, annotations = annotations, genome = genome.fa, quant = FALSE)7. Количественно оцениваются только известные трансгены и гены, новые транскрипты не идентифицируются.
se.quantOnly <- bambu(reads = samples, annotations = annotations, genome = genome.fa, discovery = FALSE)8. Сборка транскрипта независимо от ссылочной аннотации.
novelAnnotations <- bambu(reads = test.bam, annotations = NULL, genome = fa.file, NDR = 0.5, quant = FALSE)Для настройки конкретных параметров и отладки обратитесь к официальному GitHub.
4. Выходные файлы
Бамбу вернётSummarizedExperimentОбъект, доступ к которому можно получить через:
- assays(se) возвращает матрицу распространенности экспрессии транскриптов в форме количества или CPM.
- rowRanges(se) возвращает список GRangesList, содержащий все аннотированные и вновь обнаруженные транскрипты.
- rowData(se) возвращает дополнительную информацию для каждой расшифровки.
Извлеките выражение транскрипта, используя переменные (например, количество или CPM) в assays().:
- assays(se)$counts - количество подсчитанных выражений.
- assays(se)$CPM — экспрессия, нормализованная по глубине секвенирования.
- assays(se)$fullLengthCounts — выражение подсчета полноразмерных последовательностей каждого транскрипта.
- assays(se)$uniqueCounts — подсчет экспрессии уникальных последовательностей для каждого транскрипта.
Можно использоватьwriteBambuOutput()Выведите полный файл результатов. Эта команда может создать три файла: 1. Расширенный..gtfдокумент. 2. Подсчитайте матрицу экспрессии транскриптов. 3. Матрица экспрессии количества генов。
writeBambuOutput(se, path = "./bambu/")Если вас интересуют только новые расшифровки, вы можете отфильтровать справочные аннотированные расшифровки.
se.novel = se[mcols(se)$novelTranscript,]
writeBambuOutput(se.novel, path = "./bambu/")5. Визуализация
проходитьplotBambuГены/транскрипты можно визуализировать (рис. 5).。
plotBambu(se, type = "annotation", gene_id = "ENSG00000107104")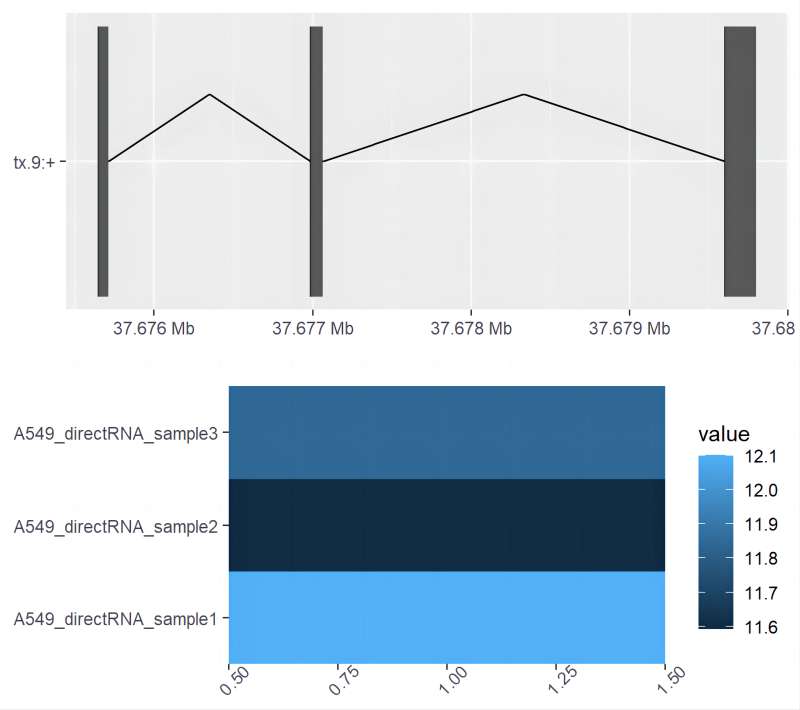
Структура и уровни экспрессии транскрипта tx.9 и других транскриптов соответствующих ему генов (рис. 6)。
plotBambu(se, type = "annotation", transcript_id = "tx.9")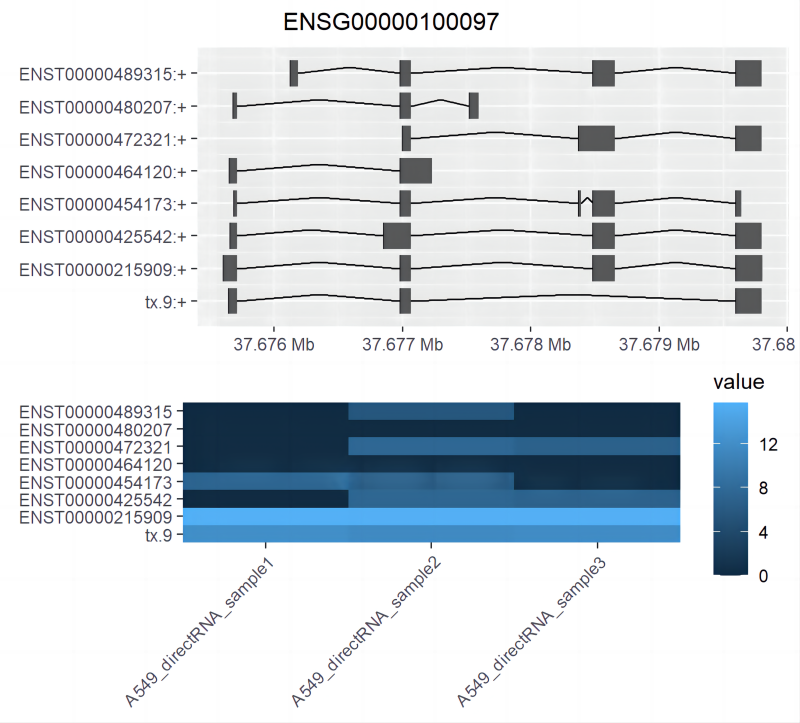
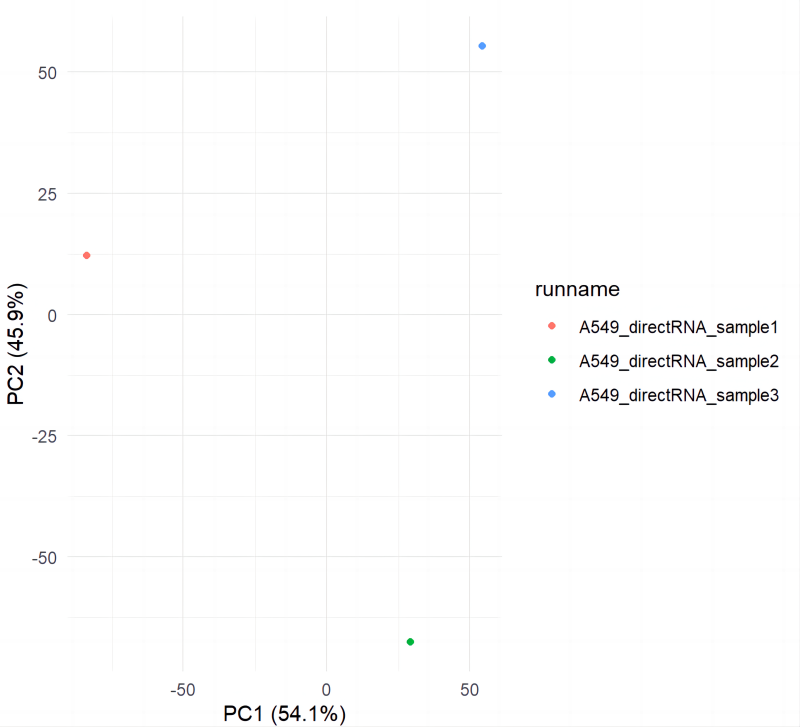
проходитьplotBambuОтображение примера группировки кластеров PCA (рис. 7)。
Ссылки
- Chen, Ying, et al. "Context-aware transcript quantification from long-read RNA-seq data with Bambu." Nature methods. (2023)
- Bambu github: https://github.com/GoekeLab/bambu



Неразрушающее увеличение изображений одним щелчком мыши, чтобы сделать их более четкими артефактами искусственного интеллекта, включая руководства по установке и использованию.
Копикодер: этот инструмент отлично работает с Cursor, Bolt и V0! Предоставьте более качественные подсказки для разработки интерфейса (создание навигационного веб-сайта с использованием искусственного интеллекта).
Новый бесплатный RooCline превосходит Cline v3.1? ! Быстрее, умнее и лучше вилка Cline! (Независимое программирование AI, порог 0)

Разработав более 10 проектов с помощью Cursor, я собрал 10 примеров и 60 подсказок.
Я потратил 72 часа на изучение курсорных агентов, и вот неоспоримые факты, которыми я должен поделиться!

Идеальная интеграция Cursor и DeepSeek API
DeepSeek V3 снижает затраты на обучение больших моделей
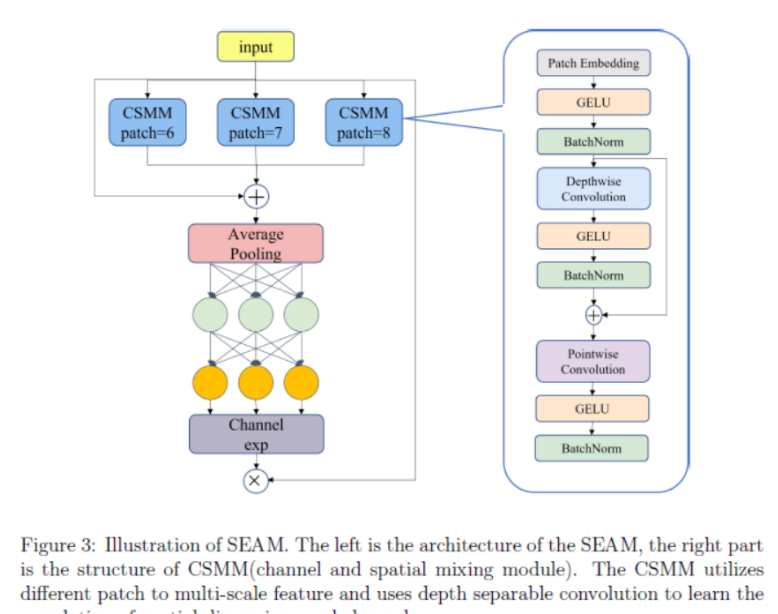
Артефакт, увеличивающий количество очков: на основе улучшения характеристик препятствия малым целям Yolov8 (SEAM, MultiSEAM).

DeepSeek V3 раскручивался уже три дня. Сегодня я попробовал самопровозглашенную модель «ChatGPT».

Open Devin — инженер-программист искусственного интеллекта с открытым исходным кодом, который меньше программирует и больше создает.
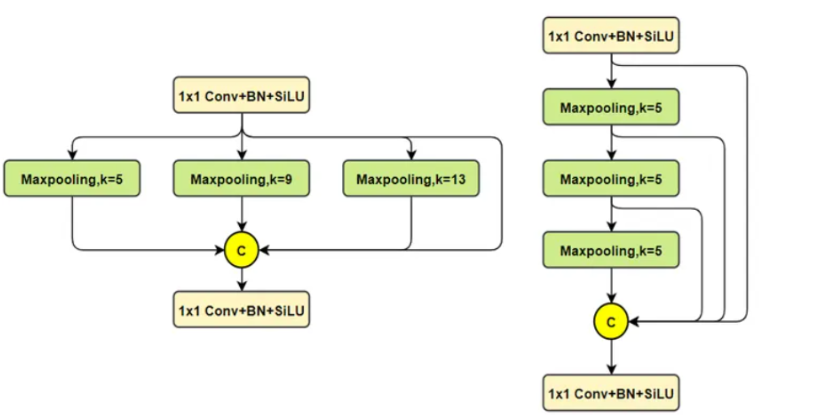
Эксклюзивное оригинальное улучшение YOLOv8: собственная разработка SPPF | SPPF сочетается с воспринимаемой большой сверткой ядра UniRepLK, а свертка с большим ядром + без расширения улучшает восприимчивое поле

Популярное и подробное объяснение DeepSeek-V3: от его появления до преимуществ и сравнения с GPT-4o.

9 основных словесных инструкций по доработке академических работ с помощью ChatGPT, эффективных и практичных, которые стоит собрать
Вызовите deepseek в vscode для реализации программирования с помощью искусственного интеллекта.
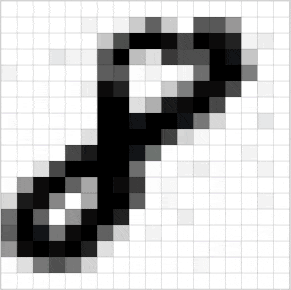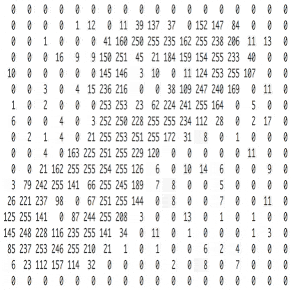
Познакомьтесь с принципами сверточных нейронных сетей (CNN) в одной статье (суперподробно)
50,3 тыс. звезд! Immich: автономное решение для резервного копирования фотографий и видео, которое экономит деньги и избавляет от беспокойства.
Cloud Native|Практика: установка Dashbaord для K8s, графика неплохая
Краткий обзор статьи — использование синтетических данных при обучении больших моделей и оптимизации производительности
MiniPerplx: новая поисковая система искусственного интеллекта с открытым исходным кодом, спонсируемая xAI и Vercel.
Конструкция сервиса Synology Drive сочетает проникновение в интрасеть и синхронизацию папок заметок Obsidian в облаке.
Центр конфигурации————Накос
Начинаем с нуля при разработке в облаке Copilot: начать разработку с минимальным использованием кода стало проще

[Серия Docker] Docker создает мультиплатформенные образы: практика архитектуры Arm64

Обновление новых возможностей coze | Я использовал coze для создания апплета помощника по исправлению домашних заданий по математике

Советы по развертыванию Nginx: практическое создание статических веб-сайтов на облачных серверах
Feiniu fnos использует Docker для развертывания личного блокнота Notepad

Сверточная нейронная сеть VGG реализует классификацию изображений Cifar10 — практический опыт Pytorch
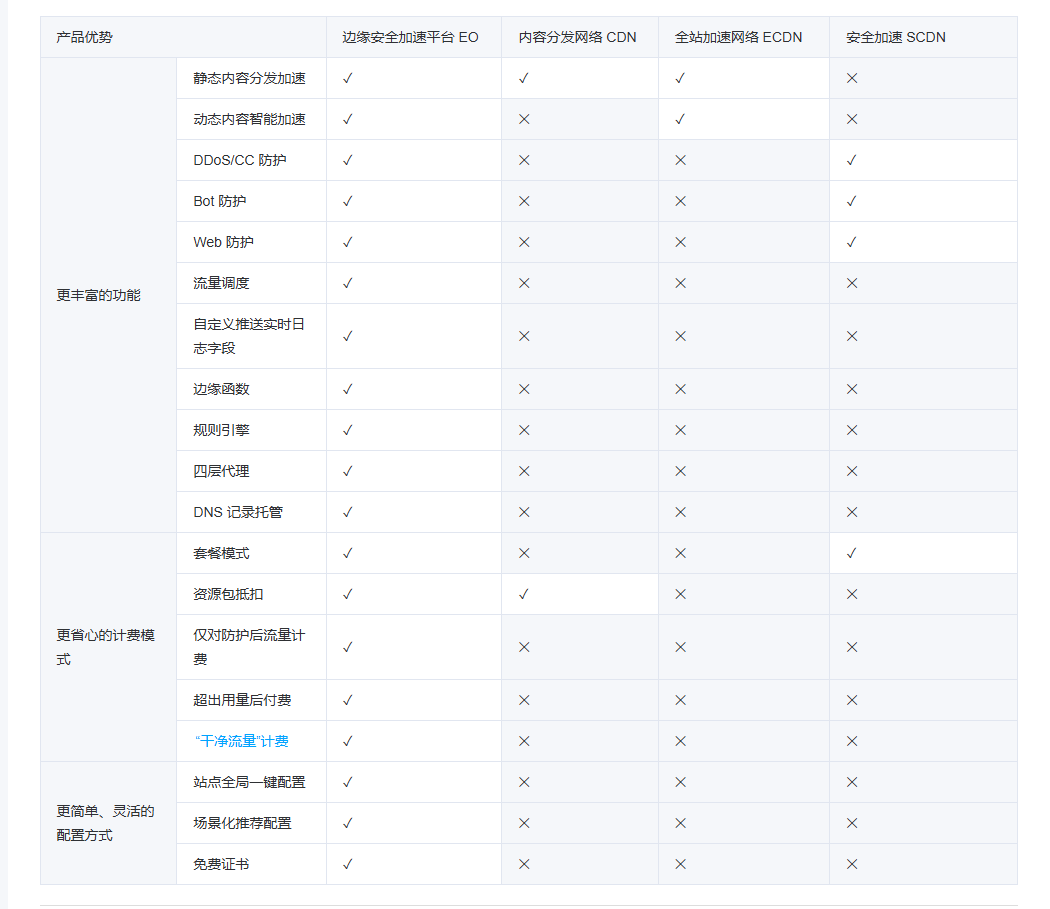
Начало работы с EdgeonePages — новым недорогим решением для хостинга веб-сайтов
[Зона легкого облачного игрового сервера] Управление игровыми архивами