Поймите определение, механизм производства и метод идентификации слияния генов в одной статье [легко понять]
Всем привет, мы снова встретились, я ваш друг Цюаньчжаньцзюнь.
[Определение слияния генов]
Слияние генов означает слияние части или всех последовательностей двух разных генов вследствие какого-либо механизма (например, мутации генома) с образованием нового гена. Как показано ниже:

Принципиальная схема слияния генов(Источник изображения:https://www.tumorfusions.org/)
Вообще говоря, слияние генов означает слияние на уровне генома. Однако слияние может происходить и на уровне транскриптома, главным образом потому, что РНК, образующиеся в результате транскрипции двух разных генов, по какой-то причине сливаются вместе, образуя новую слитую РНК, которая может кодировать белок или может быть некодирующей. Гены слияния, генерируемые на уровне генома, могут экспрессироваться или не экспрессироваться в зависимости от ситуации слияния (например, повреждения области промотора или других причин).
[Механизм слияния генов]
Существует три основных механизма слияния генов, как показано на рисунке ниже:

Три распространенных механизма слияния генов(Источник изображения:Wikipedia)
Существует три общих механизма слияния генов:
1) Хромосомная транслокация, хромосомная транслокация. Как показано на рисунке А выше, два сегмента хромосом 1 и 2 пересекаются и обмениваются местами, в результате чего светло-зеленый ген на хромосоме 1 сливается с оранжевым геном на хромосоме 2;
2) Интерстициальная делеция, отсутствует середина. Как показано на рисунке выше, сегмент между оранжевым геном и светло-зеленым геном на хромосоме 3 удаляется, что в конечном итоге приводит к слиянию двух генов;
3) Хромосомная инверсия, хромосомная инверсия. Например, сегмент между геном оранжевого и геном темно-зеленого цвета на хромосоме 4 подвергается инверсии, что в конечном итоге приводит к слиянию гена оранжевого и гена светло-зеленого.
[Связь между слиянием генов и раком]
Так зачем же изучать слияние генов? Поскольку многие прошлые исследования постоянно показывали, что слияние генов тесно связано с возникновением и развитием различных заболеваний, особенно рака, и даже является прямой причиной некоторых видов рака, слияние генов также стало важным исследованием в современном анализе больших данных омикса. . содержание.
В настоящее время сообщается, что возникновение многих видов рака тесно связано со слиянием генов, как показано в следующей таблице:

Количество известных слияний генов и количество повторных слияний в некоторых опухолях (Изображение предоставлено: Мертенс et al. Nature Reviews Cancer, 2015)
Более того, FDA США (Управление по контролю за продуктами и лекарствами) одобрило некоторые препараты, нацеленные на определенные слияния генов, для лечения соответствующих видов рака, как показано в следующей таблице:

Одобренные FDA препараты для лечения соответствующих опухолей ( Источник изображения: Мертенс et al. Nature Reviews Cancer, 2015)
Следовательно, слияния генов могут быть тесно связаны с возникновением и развитием различных видов рака. Эти слитые гены также могут быть потенциальными мишенями для лекарств, и необходимо провести их углубленное исследование.
[Идентификация слияний генов на основе полногеномного секвенирования и секвенирования транскриптома]
Идентификация слияний генов может быть основана на данных полногеномного секвенирования (WGS), данных секвенирования транскриптома (RNA-seq), или лучше использовать комбинацию этих двух технологий.
Слияние генов, выявленное с помощью полногеномного секвенирования, в основном можно определить как вызванное какой-либо мутацией на уровне генома. Однако без данных секвенирования транскриптома невозможно точно определить, может ли новый ген, созданный после слияния, экспрессироваться или нет. количество выражения. Высокое и низкое.
Слияние генов, выявленное по данным секвенирования транскриптома, может быть четко выражено как слияние генов, но невозможно полностью определить, вызвано ли оно геномной вариацией или происходит в результате слияния РНК, которое происходит после транскрипции двух разных генов.
Следовательно, если позволяют условия, более точные результаты идентификации могут быть получены путем объединения полногеномного секвенирования и секвенирования транскриптома для выявления слияний генов.
[Общие термины при идентификации слияния генов]
Прежде чем разобраться в методах или программном обеспечении для идентификации слияний генов, давайте сначала разберемся с некоторыми общими терминами, используемыми при идентификации слияний генов на основе данных секвенирования. Подробности показаны на рисунке ниже:

Некоторые общие термины для идентификации слияния генов(Источник изображения:Liu et al. Nucleic Acids Research, 2016)(A) Intact exon (IE) type andbroken exon (BE) type fusion transcripts; (B) spanning read, split readand anchor length; (C) short and long insert size of DNA fragment forsequencing.
Эти общие термины:
1) Слияние типа интактного экзона (IE) означает, что исходный экзон полностью сохраняется после слияния, не затрагивая исходную структуру экзона. Как показано на рисунке A выше, слияние экзона 2 гена A и экзона 1 гена B полностью сохраняет последовательность двух экзонов;
2) Слияние типа сломанного экзона (BE) означает, что исходная полная последовательность экзонов не сохраняется после слияния. Как показано на рисунке A выше, частичная последовательность экзона 3 гена A слита с экзоном 2 гена B. В новом гене после слияния часть последовательности экзона 3 гена A теряется;
3) Точка разрыва относится к месту слияния двух слитых генов в геноме, например, к месту слияния гена A (синий) и гена B (зеленый) на рисунке B выше;
4) Связующее чтение относится к парному чтению, которое соответствует двум слитым генам в сайте слияния, например, пара ридов, соответствующих гену A (синий) и гену B (зеленый) на рисунке B выше;
5) Разделенное чтение относится к прочтению, которое точно соответствует месту слияния, как показано в правой части рисунка B выше;
6) Длина якоря относится к длине левого и правого концов считывания, охватывающего место сращения, как показано на правой стороне рисунка B выше;
7) Короткий размер вставки обычно относится к короткому расстоянию между двумя прочтениями при секвенировании парных концов, которое обычно составляет несколько сотен пар оснований;
8) Длинный размер вставки обычно относится к большему расстоянию между двумя прочтениями при секвенировании парных концов, которое обычно составляет несколько килобайт или даже больше;
Разработка программного обеспечения для идентификации слияния генов обычно разрабатывается на основе упомянутых выше условий и с использованием соответствующих алгоритмов.
[Сравнение производительности программного обеспечения для идентификации слияния генов]
На данный момент разработаны десятки различных программ для обнаружения слияния генов, некоторые из которых имеют относительно хорошие комплексные характеристики. Далее мы продолжим сравнивать и анализировать производительность некоторых широко используемых программ для идентификации слияния генов.
В следующей таблице приведено сравнение производительности 15 широко используемых программ для идентификации слияния генов на 3 различных типах синтетических данных и 3 наборах реальных данных. Это программное обеспечение: SOAPfuse, FusionCatcher, JAFFA, EricScript, chimerascan, PRADA, deFuse, FusionMap, TopHat-Fusion, MapSplice, BreakFusion, SnowShoes-FTD, FusionQ, FusionHunter, ShortFuse.

Сравнение показателей F-мер 15 типов программного обеспечения для идентификации слияния генов на 3 наборах синтетических данных и 3 наборах реальных данных(Источник изображения:Liu et al. Nucleic Acids Research, 2016). F-мера — это статистика, также известная как F-показатель, которая представляет собой средневзвешенное гармоническое значение точности и полноты. Она часто используется для оценки качества моделей классификации. Чем выше значение, тем лучше производительность. Примечание:* Лучшая общая производительность.
Стоит отметить, что длина считываний секвенирования, а также размер вставки считываний парного секвенирования и т. д. будут влиять на идентификационный эффект слияния генов. Поэтому в приведенной выше таблице используются различные типы данных измерений для всестороннего изучения производительности этих 15 программ. Среди них Тип-1А представляет собой данные секвенирования парных концов химерных транскриптов с 5'- и 3'-конца, искусственно синтезированных с использованием программного обеспечения wgsim, где длина считывания составляет 100 п.о., а размер вставки - 500 ± 50 п.о. Синтез данных типа 1B; метод и Тип-1А аналогичны, за исключением того, что размер вставки данных парного секвенирования меньше, 250 ± 50 п.н. Тип-3B: синтетические данные с длиной считывания 50 п.н.; Остальные три набора реальных данных относятся к раку молочной железы, меланоме и раку простаты соответственно.
Результаты сравнения показывают, что SOAPfuse, FusionCatcher и JAFFA имеют лучшую общую производительность на 3 наборах смоделированных данных и 3 наборах реальных данных и достигли наивысшего балла по F-мере.
Лю и соавт. далее сравнили эффективность программного обеспечения для обнаружения слияния 15 генов на более реальных данных. Подробности показаны на рисунке ниже:

Дальнейшее сравнение производительности программного обеспечения для обнаружения слияния 15 генов на реальных наборах данных секвенирования.(Источник изображения: Liu et al. Nucleic Acids Research, 2016 ). Вертикальная ось от A до C — количество обнаруженных реальных слияний генов, а горизонтальная ось — 15 различных программ. На рисунке D показано сравнение кривых между Precision и Recall. Среди них (А) и (D) использует Breast набор данных о раке; (B) и (E) Используется набор данных меланомы; (C) и(F) использует простату набор данных о раке.
Результаты сравнения с реальными данными также показывают, что SOAPfuse, FusionCatcher и JAFFA имеют более высокую точность обнаружения слияний генов.
Liu et al. Также сравнивалось время работы программного обеспечения для обнаружения слияния 15 генов на синтетических наборах данных и реальных наборах данных с различной глубиной секвенирования. Подробности показаны на рисунке ниже:

Сравнение скорости работы программного обеспечения для обнаружения слияния 15 генов(Источник изображения: Liu et al. Nucleic Acids Research, 2016 ). По оси Y отложено время работы в минутах (мин). А есть Синтетический набор данных, длина чтения 100. п.н., глубина моделирования секвенирования составляет 50X соответственно, 100X и200X. Б – недвижимость cancer 171T наборов данных.
Результаты сравнения времени работы показывают, что такое программное обеспечение, как FusionMap, работает быстрее всего. Однако предыдущие результаты показывают, что точность слияния генов, обнаруженного с помощью FusionMap, низка.
так,Ни один метод не имеет наиболее очевидных преимуществ во всех аспектах сравнения производительности. общий,SOAPfuse — лучшее общее сравнение,Далее идет FusionCatcher и JAFFA. Более того, поскольку разные программы имеют разные преимущества и недостатки, более точные результаты можно получить, объединив несколько разных программ для выявления слияний генов.
Издатель: Лидер стека программистов полного стека, укажите источник для перепечатки: https://javaforall.cn/166940.html Исходная ссылка: https://javaforall.cn



Неразрушающее увеличение изображений одним щелчком мыши, чтобы сделать их более четкими артефактами искусственного интеллекта, включая руководства по установке и использованию.
Копикодер: этот инструмент отлично работает с Cursor, Bolt и V0! Предоставьте более качественные подсказки для разработки интерфейса (создание навигационного веб-сайта с использованием искусственного интеллекта).
Новый бесплатный RooCline превосходит Cline v3.1? ! Быстрее, умнее и лучше вилка Cline! (Независимое программирование AI, порог 0)

Разработав более 10 проектов с помощью Cursor, я собрал 10 примеров и 60 подсказок.
Я потратил 72 часа на изучение курсорных агентов, и вот неоспоримые факты, которыми я должен поделиться!

Идеальная интеграция Cursor и DeepSeek API
DeepSeek V3 снижает затраты на обучение больших моделей
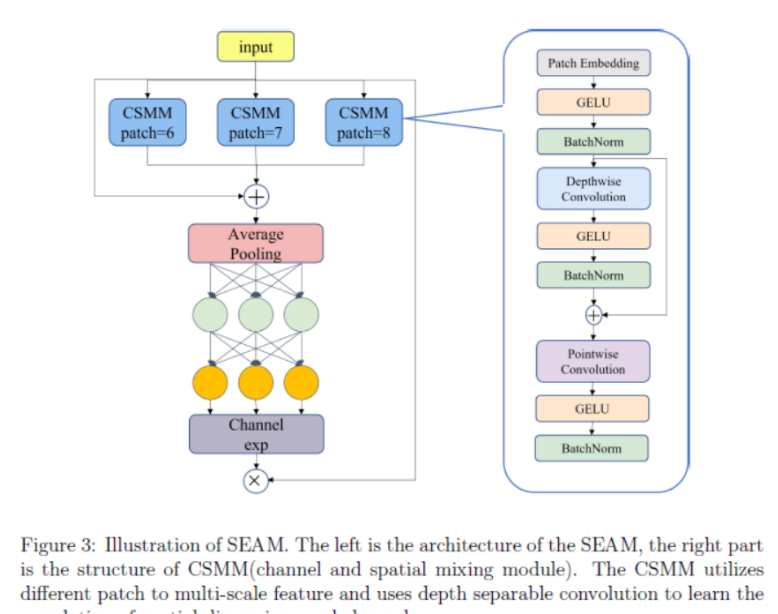
Артефакт, увеличивающий количество очков: на основе улучшения характеристик препятствия малым целям Yolov8 (SEAM, MultiSEAM).

DeepSeek V3 раскручивался уже три дня. Сегодня я попробовал самопровозглашенную модель «ChatGPT».

Open Devin — инженер-программист искусственного интеллекта с открытым исходным кодом, который меньше программирует и больше создает.
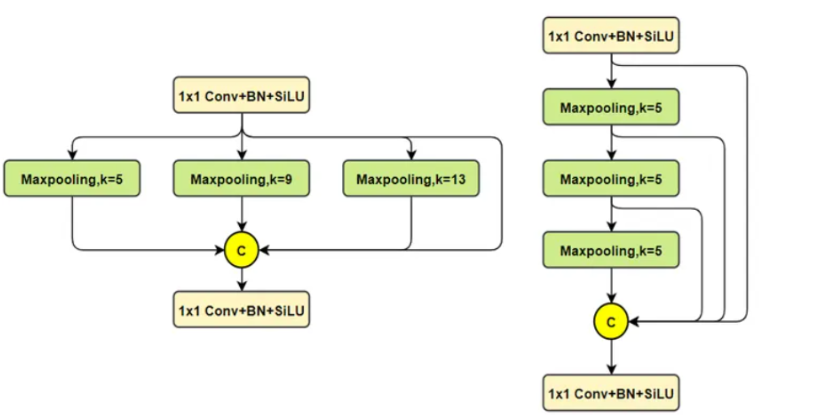
Эксклюзивное оригинальное улучшение YOLOv8: собственная разработка SPPF | SPPF сочетается с воспринимаемой большой сверткой ядра UniRepLK, а свертка с большим ядром + без расширения улучшает восприимчивое поле

Популярное и подробное объяснение DeepSeek-V3: от его появления до преимуществ и сравнения с GPT-4o.

9 основных словесных инструкций по доработке академических работ с помощью ChatGPT, эффективных и практичных, которые стоит собрать
Вызовите deepseek в vscode для реализации программирования с помощью искусственного интеллекта.
Познакомьтесь с принципами сверточных нейронных сетей (CNN) в одной статье (суперподробно)
50,3 тыс. звезд! Immich: автономное решение для резервного копирования фотографий и видео, которое экономит деньги и избавляет от беспокойства.
Cloud Native|Практика: установка Dashbaord для K8s, графика неплохая
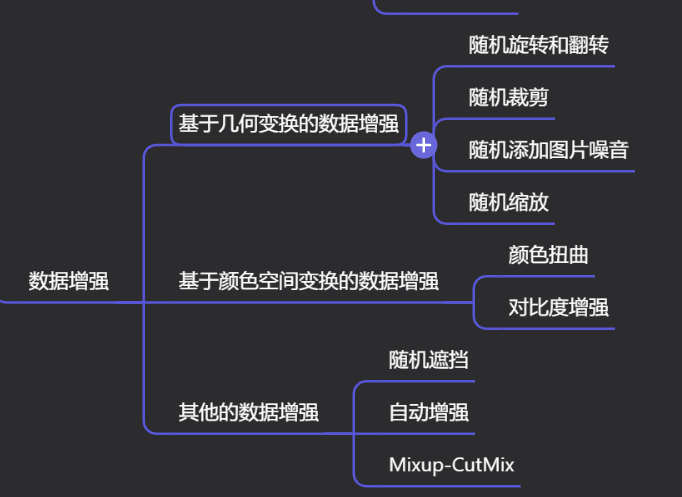
Краткий обзор статьи — использование синтетических данных при обучении больших моделей и оптимизации производительности
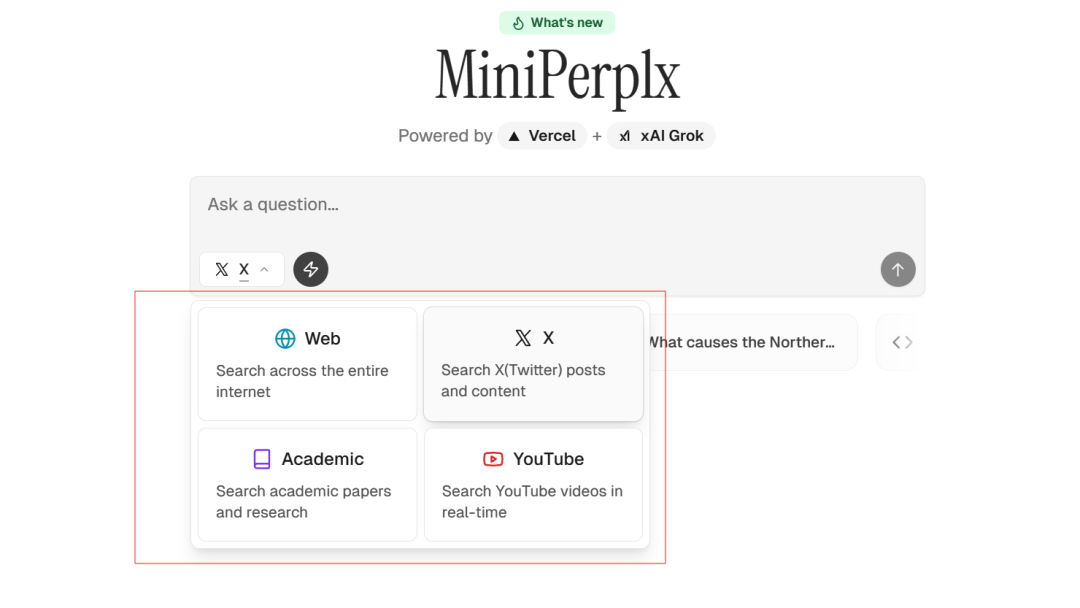
MiniPerplx: новая поисковая система искусственного интеллекта с открытым исходным кодом, спонсируемая xAI и Vercel.
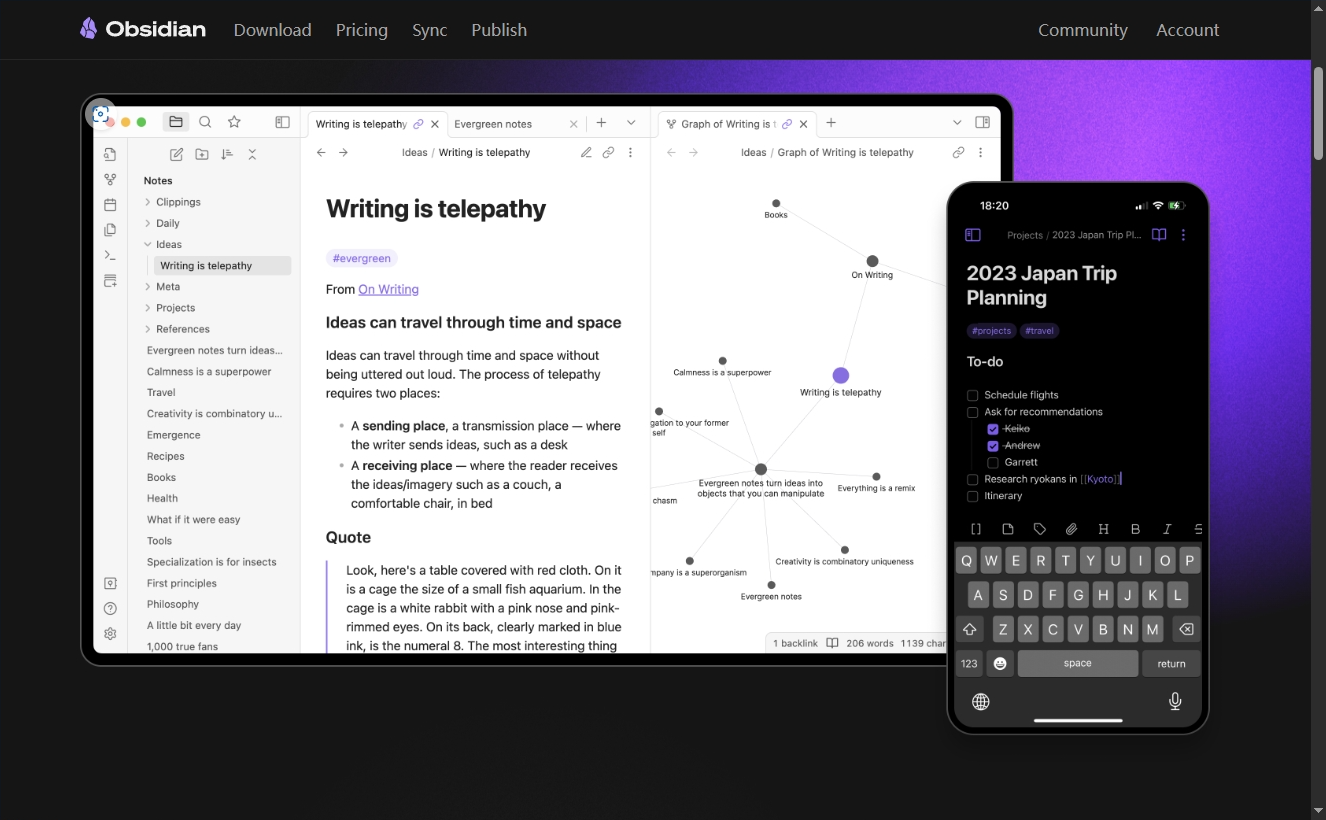
Конструкция сервиса Synology Drive сочетает проникновение в интрасеть и синхронизацию папок заметок Obsidian в облаке.
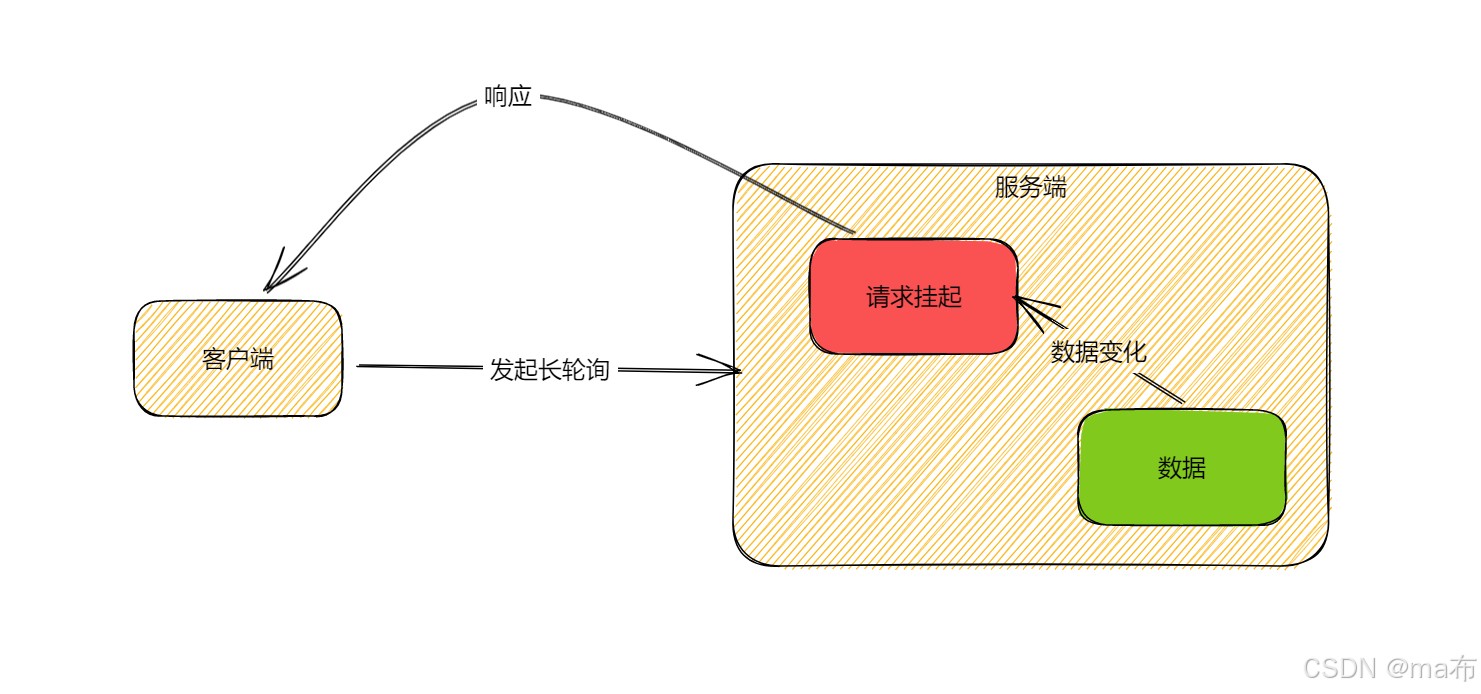
Центр конфигурации————Накос
Начинаем с нуля при разработке в облаке Copilot: начать разработку с минимальным использованием кода стало проще

[Серия Docker] Docker создает мультиплатформенные образы: практика архитектуры Arm64

Обновление новых возможностей coze | Я использовал coze для создания апплета помощника по исправлению домашних заданий по математике

Советы по развертыванию Nginx: практическое создание статических веб-сайтов на облачных серверах
Feiniu fnos использует Docker для развертывания личного блокнота Notepad

Сверточная нейронная сеть VGG реализует классификацию изображений Cifar10 — практический опыт Pytorch
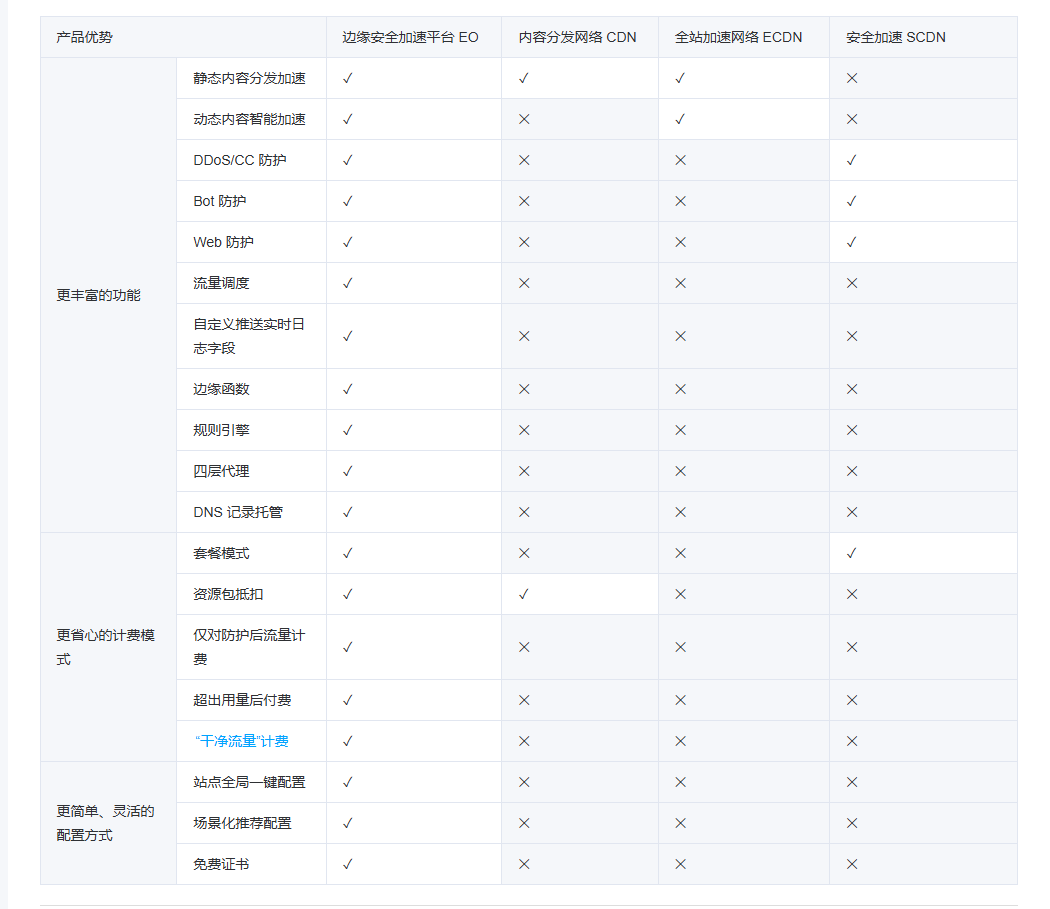
Начало работы с EdgeonePages — новым недорогим решением для хостинга веб-сайтов
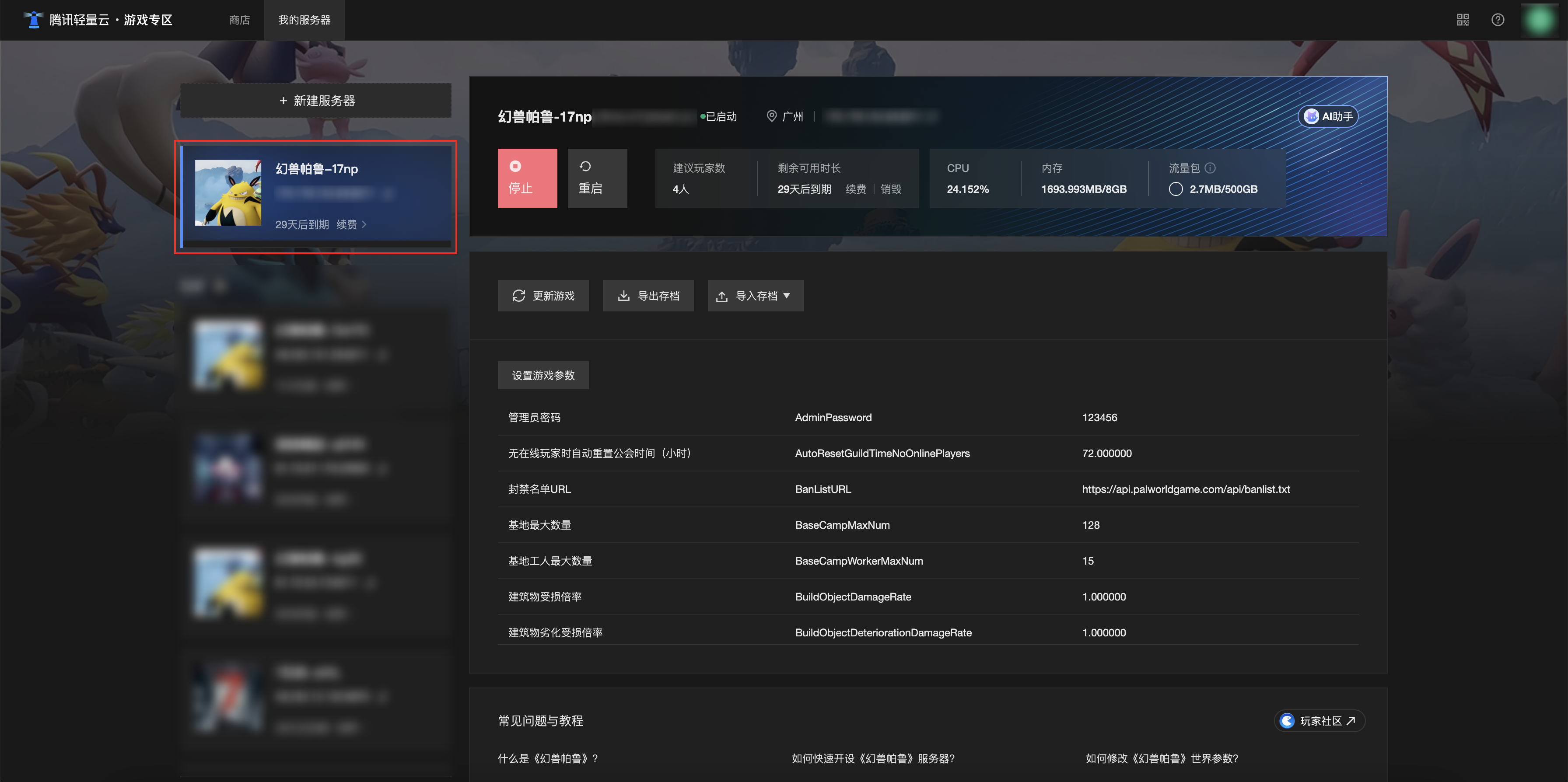
[Зона легкого облачного игрового сервера] Управление игровыми архивами