Поделитесь способом быстрой загрузки наборов данных SRA
Всем привет! Все мы знаем, что при проведении биоинформатического анализа,Будет использован исходный файл datafastq. но,Когда мы хотим использовать данные секвенирования других людей для повторного анализа, мы обычно не можем напрямую загрузить файл fastq из базы данных NCBI, а должны сначала загрузить данные SRA.Так,Как эффективно скачать СРАданные?,На данный момент основные методы включают 5 типов: SRA от NCBI Официально доступен Загрузите набор инструментов прямо по ссылке или загрузите его с помощью wget в Linux с помощью aspera; Высокоскоростная загрузка; используйте Grabseqs; загрузка инструмента; использование сканера Python и других инструментов может помочь вам загрузить его.Многие друзья написали об этих методах.,Просто спросите напрямую у Ду Ньянга, какой метод вы хотите использовать!
База данных SRA (Sequence Read Archive) — это дополнительная библиотека NCBI (Национального центра биотехнологической информации), специально используемая для хранения данных высокопроизводительного секвенирования. База данных SRA, в которой собраны необработанные данные секвенирования со всего мира, которые можно загрузить бесплатно, является ценным ресурсом для исследователей в области медико-биологических наук.
тип данных
Типы данных хранилища данных SRA включают:
- Необработанные данные секвенирования (необработанные sequencing данные), файл fastq
- Информация о выравнивании последовательностей (информация о выравнивании), файл bam
Загрузка данных
Многие журналы требуют от авторов публично публиковать данные высокопроизводительного секвенирования в SRA при публикации статьи. Данные, загружаемые в SRA, требуют определенных процессов подготовки и загрузки, в том числе:
- Установите программное обеспечение FTP (например, FileZilla) для загрузки данных.
- Подготовьте файлы данных. Распространенные типы файлов включают fasta, fastq, bam и т. д.
- Загрузка данных через Портал подачи библиотеки данных SRA.
Загрузка данных
Исследователи могут загружать данные о последовательностях из базы данных SRA различными способами, в том числе:
- Загрузка веб-страницы
- Используйте плагин для браузера (например, Aspera Connect)
- Загрузите с помощью пакета SRAинструмент.
структура данных
Структура данных библиотеки данных SRA построена на следующих четырех концепциях:
- ИЗУЧЕНИЕ: тема исследования/проект с такими префиксами, как SRP, DRP и ERP.
- SAMPLE: образец информации, префикс, например SRS, DRS, ERS.
- ЭКСПЕРИМЕНТ: Информация об эксперименте. Эксперимент может содержать несколько образцов и RUN с такими префиксами, как SRX, DRX и ERX.
- RUN: соответствует определенному циклу секвенирования с такими префиксами, как SRR, DRR и ERR.
интеллектуальный анализ данных
Данные из базы данных SRA могут использоваться для различных исследовательских целей, включая, помимо прочего:
- Найдите исходные данные опубликованной статьи.
- Изучите некодирующие гены и регуляторные элементы ДНК.
- Изучите транссплайсинг и кольцевые РНК.
- Анализ de novo необработанных данных секвенирования,Откройте для себя новые гены.
База данных SRA предоставляет научным исследователям мощный ресурс данных, который помогает способствовать прогрессу исследований в области наук о жизни.
Сегодня мы хотим поделиться с вами еще одним SRA, который, по нашему мнению, является более удобным, применимым и быстрым. Ключом к загрузке набора данных является то, что его можно использовать бесплатно.(на самом деле,Это способ загрузить программное обеспечение idm прямо по ссылке)! Если вам понравилось, не забудьте сохранить! Потому что операция проста,Итак, перейдем непосредственно к делу:
Шаг 1. Откройте NCBI и введите набор данных PRJNA778726 (образец набора данных), который будет загружен в рамках SRA.
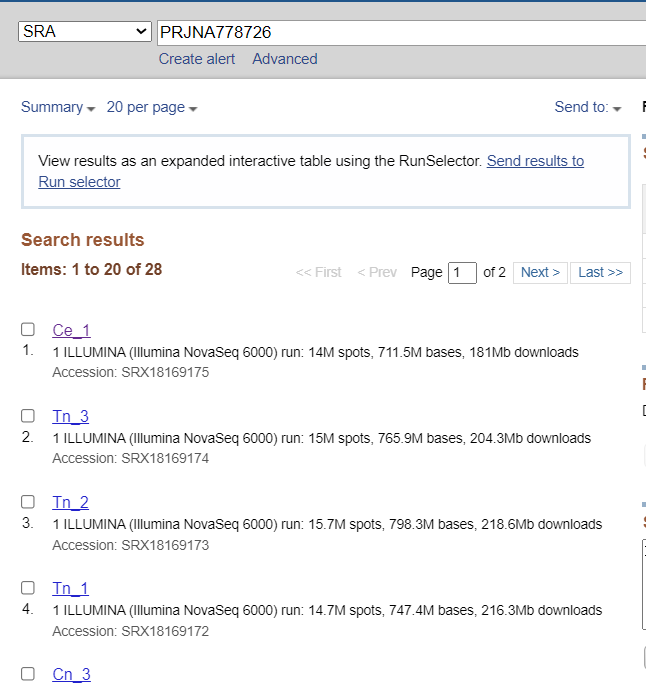
Шаг 2. Нажмите «Отправить» и выберите «Файл» → «Выполнить» → «Создать файл».
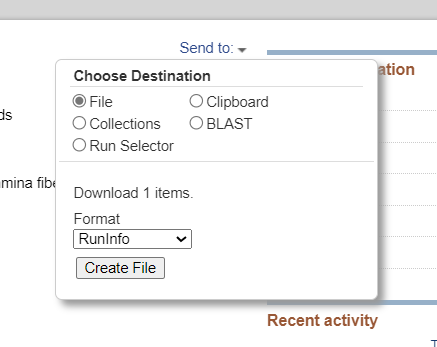
Шаг 3. После того, как данные Загрузки поступают локально, мы открываем их в Excel.
Вы обнаружите, что эта таблица содержит много контента. Обычно мы фокусируемся на следующих столбцах: Run, Download_path, ExperimenterLibraryName, LibraryStrategy, LibraryLayout, Platform, ScientificName, SampleName и Sex и т. д. Вам нужно найти ссылку для скачивания внутри.
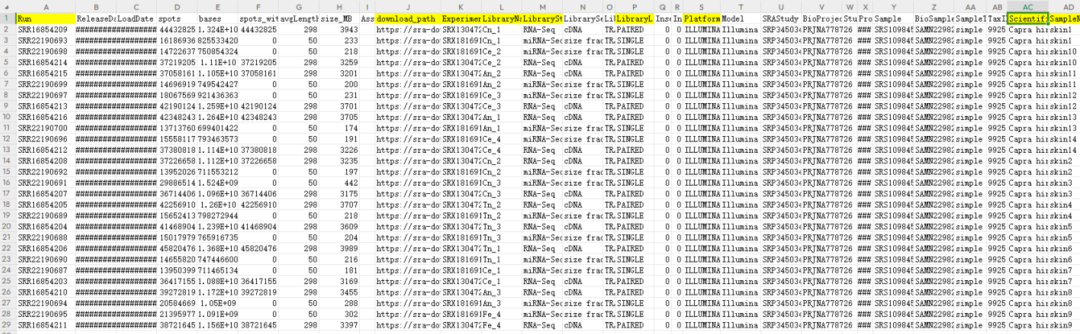
Шаг 4. На основе приведенной выше информации выберите образец данных, подходящий для вашей темы исследования, скопируйте путь загрузки в программное обеспечение IDM и загрузите его с точками останова.
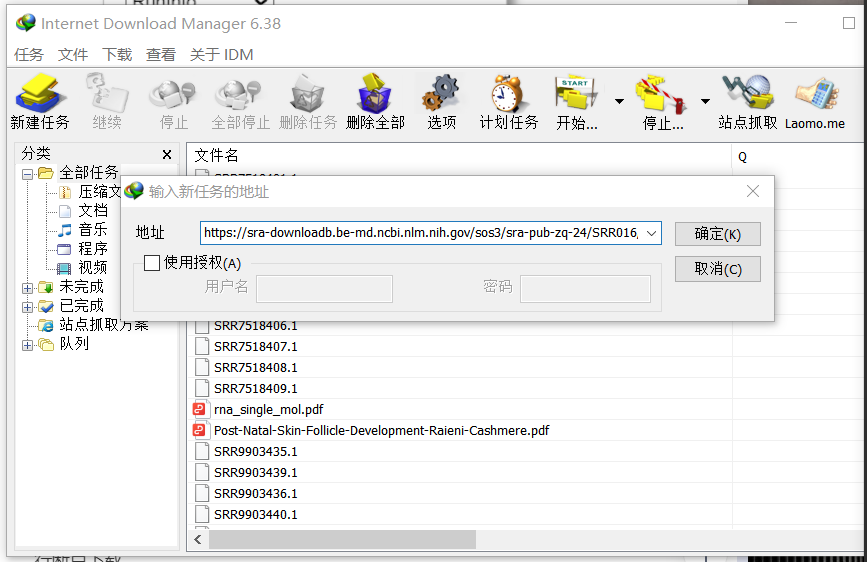
Шаг 5. Проверьте результаты и продолжительность загрузки, поддержите загрузку точки останова, очень быстро!
ХОРОШО! Вот и все, что я хочу сегодня рассказать. Содержание очень простое. Надеюсь, мой небольшой рассказ сможет пролить немного света на ваш путь научных исследований!
В конце статьи я рекомендую всем изучить базу данных SRA (Sequence Read Archive), которая предоставляет ряд API (интерфейсов прикладного программирования).,Позволяет исследователям и разработчикам программно получать доступ к данным в SRA и манипулировать ими. Ниже приведены некоторые часто используемые API базы данных SRA:
- E-utilities API:NCBIпредоставилE-utilities API, который позволяет пользователям получать данные из различных библиотек данных NCBI, включая SRA. Пользователи могут получить определенные наборы SRA, написав скрипт, например, через Run. Accession Поиск чисел.
- SRA Toolkit:SRA Toolkit представляет собой набор для скачивания.、иметь дело сипроверятьхранилищесуществоватьNCBIсеквенирование следующего поколения вданныеизинструмент。Он включает в себя несколько командных строкинструмент,нравиться
prefetchиfastq-dump,Этот инструмент можно использовать для автоматизации загрузки и конвертации SRAданных. - Aspera Connect:Aspera Connect — клиент высокоскоростной передачи данных, работающий с SRA. Интеграцию с набором инструментов можно использовать для быстрой загрузки SRAданных.
- SRA Explorer:SRA Explorer — это веб-приложение,Создан для облегчения поиска и загрузки SRAданных. Он поддерживает пользователей для поиска и выбора коллекций данных через графический интерфейс.,И может генерировать сценарии командной строки для загрузки.
- SRA API:SRAданные Библиотека Может бытьпредоставилпрямойизAPIинтерфейс,Позволяет пользователям отправлять и получать данные с помощью метода программирования. Конкретную документацию по API и методы использования можно найти на официальном сайте NCBI.
- Библиотека языков программирования:Некоторыйпрограммирование Языки могут иметь специализированныеиз Библиотекаили модуль,Используется для упрощения взаимодействия с библиотекой SRAданные.,нравитьсяPythonиз
BiopythonБиблиотека。 - Интеграция облачных сервисов:SRAданные Также доступно через несколько поставщиков облачных услуг.,Это облегчает крупномасштабный анализ.
- API отправки данных SRA:Для надеждыSRAпредставлять на рассмотрениеданныеизисследователь,NCBI предоставляет подробную информацию и рекомендации по подаче SRA.
При использовании этих API пользователи обязаны соблюдать Условия использования и Политику использования данных NCBI. Конкретные методы и параметры использования API могут со временем обновляться, поэтому для получения последней информации рекомендуется напрямую обращаться к официальной документации или ссылкам на ресурсы, предоставленные NCBI.



Неразрушающее увеличение изображений одним щелчком мыши, чтобы сделать их более четкими артефактами искусственного интеллекта, включая руководства по установке и использованию.
Копикодер: этот инструмент отлично работает с Cursor, Bolt и V0! Предоставьте более качественные подсказки для разработки интерфейса (создание навигационного веб-сайта с использованием искусственного интеллекта).
Новый бесплатный RooCline превосходит Cline v3.1? ! Быстрее, умнее и лучше вилка Cline! (Независимое программирование AI, порог 0)

Разработав более 10 проектов с помощью Cursor, я собрал 10 примеров и 60 подсказок.
Я потратил 72 часа на изучение курсорных агентов, и вот неоспоримые факты, которыми я должен поделиться!

Идеальная интеграция Cursor и DeepSeek API
DeepSeek V3 снижает затраты на обучение больших моделей
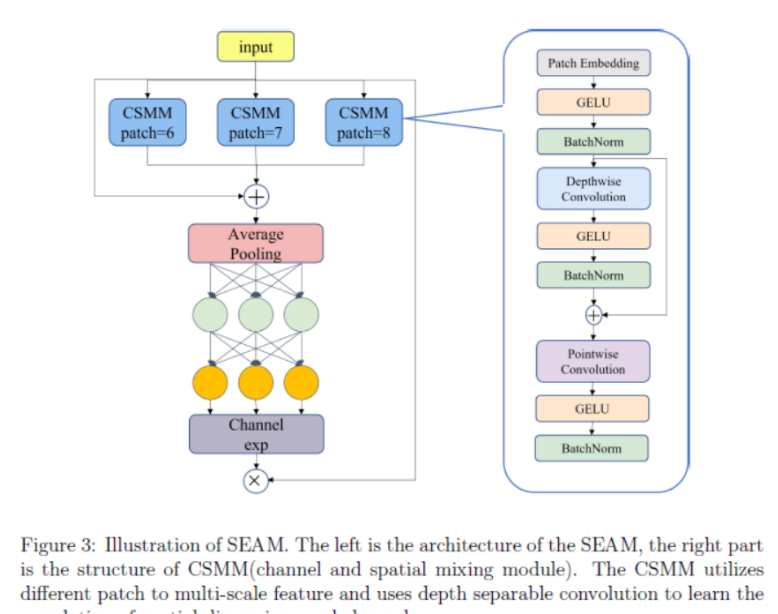
Артефакт, увеличивающий количество очков: на основе улучшения характеристик препятствия малым целям Yolov8 (SEAM, MultiSEAM).

DeepSeek V3 раскручивался уже три дня. Сегодня я попробовал самопровозглашенную модель «ChatGPT».

Open Devin — инженер-программист искусственного интеллекта с открытым исходным кодом, который меньше программирует и больше создает.
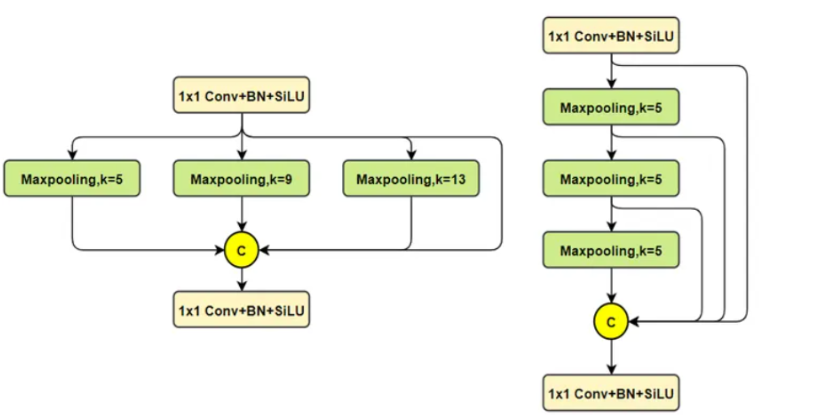
Эксклюзивное оригинальное улучшение YOLOv8: собственная разработка SPPF | SPPF сочетается с воспринимаемой большой сверткой ядра UniRepLK, а свертка с большим ядром + без расширения улучшает восприимчивое поле

Популярное и подробное объяснение DeepSeek-V3: от его появления до преимуществ и сравнения с GPT-4o.

9 основных словесных инструкций по доработке академических работ с помощью ChatGPT, эффективных и практичных, которые стоит собрать
Вызовите deepseek в vscode для реализации программирования с помощью искусственного интеллекта.
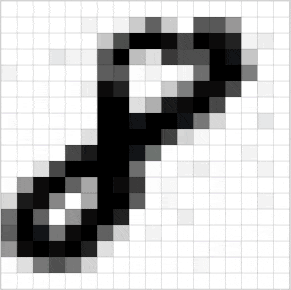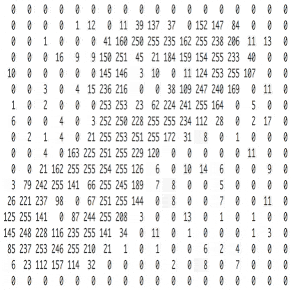
Познакомьтесь с принципами сверточных нейронных сетей (CNN) в одной статье (суперподробно)
50,3 тыс. звезд! Immich: автономное решение для резервного копирования фотографий и видео, которое экономит деньги и избавляет от беспокойства.
Cloud Native|Практика: установка Dashbaord для K8s, графика неплохая
Краткий обзор статьи — использование синтетических данных при обучении больших моделей и оптимизации производительности
MiniPerplx: новая поисковая система искусственного интеллекта с открытым исходным кодом, спонсируемая xAI и Vercel.
Конструкция сервиса Synology Drive сочетает проникновение в интрасеть и синхронизацию папок заметок Obsidian в облаке.
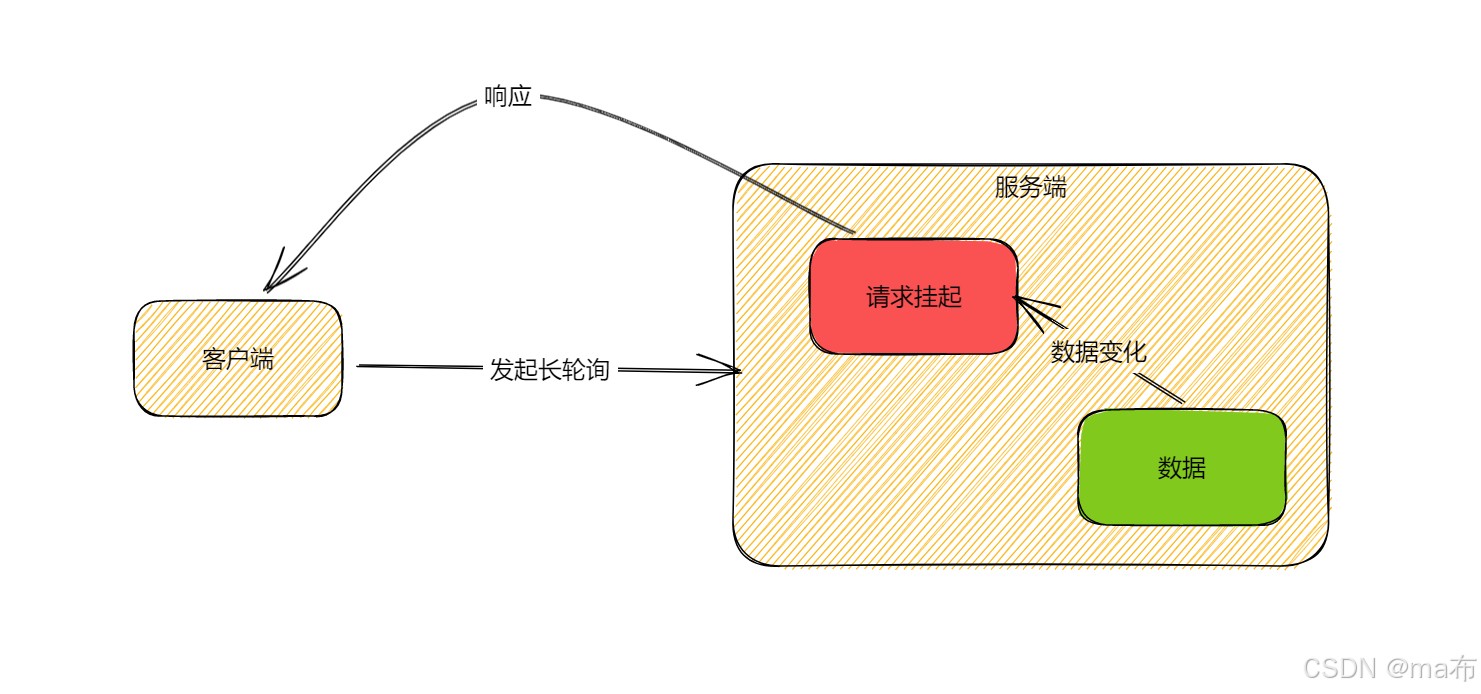
Центр конфигурации————Накос
Начинаем с нуля при разработке в облаке Copilot: начать разработку с минимальным использованием кода стало проще

[Серия Docker] Docker создает мультиплатформенные образы: практика архитектуры Arm64

Обновление новых возможностей coze | Я использовал coze для создания апплета помощника по исправлению домашних заданий по математике

Советы по развертыванию Nginx: практическое создание статических веб-сайтов на облачных серверах
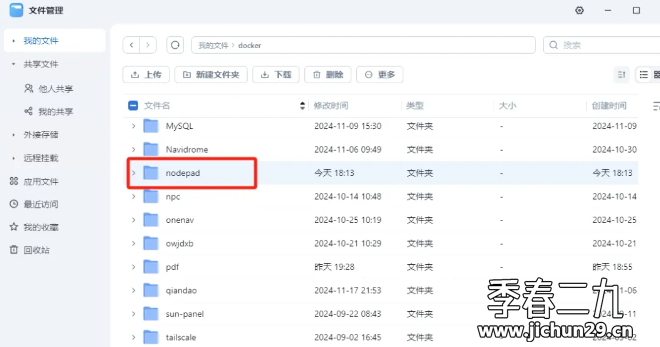
Feiniu fnos использует Docker для развертывания личного блокнота Notepad

Сверточная нейронная сеть VGG реализует классификацию изображений Cifar10 — практический опыт Pytorch
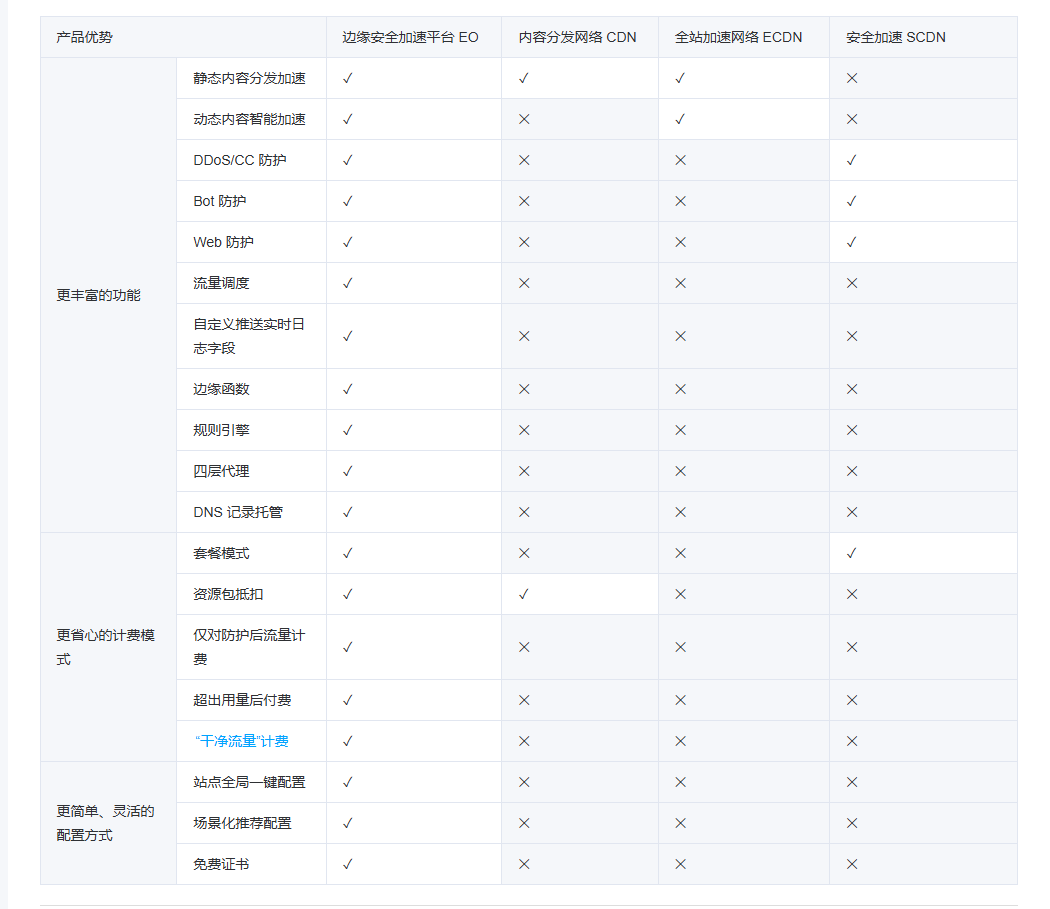
Начало работы с EdgeonePages — новым недорогим решением для хостинга веб-сайтов
[Зона легкого облачного игрового сервера] Управление игровыми архивами