Обрабатывайте данные быстрее — Sambamba
Если рабочий хочет хорошо выполнять свою работу, он должен сначала заточить свои инструменты.
1sambamba
Sambamba — эффективный инструмент биоинформатики, разработанный Артемом Тарасовым. Он в основном используется для обработки крупномасштабных данных секвенирования, особенно файлов в формате SAM/BAM. Это программное обеспечение предназначено для обеспечения более высокой производительности, чем существующие инструменты (samtools), особенно в многоядерных процессорных системах, используя преимущества многоядерной обработки и значительно сокращая время обработки. Он имеет следующие свойства:
- Многопоточность:SambambaВозможность использовать преимущества многоядерных процессоровиз Преимущества,Ускорение за счет параллельной обработкиданныеизчитать、сортировать индексацию и другие операции.
- совместимость:Sambambaдизайни Обычно используетсяизсвидетельство о рожденииинструментнравитьсяSamtoolsсовместимый,что позволяет легко интегрировать егоприезжатьсуществующийиз Процесс анализасередина。
- Основные языки программирования:Dязык
- Платформа поддержки:поддерживатьLinuxиMacOS,Не поддерживается в Windows
2Публикация статей
тема:Sambamba: fast processing of NGS alignment formats Журнал:Bioinformatics дата:2015/6/15 автор&единица:Artem Tarasov DOI:https://doi.org/10.1093/bioinformatics/btv098
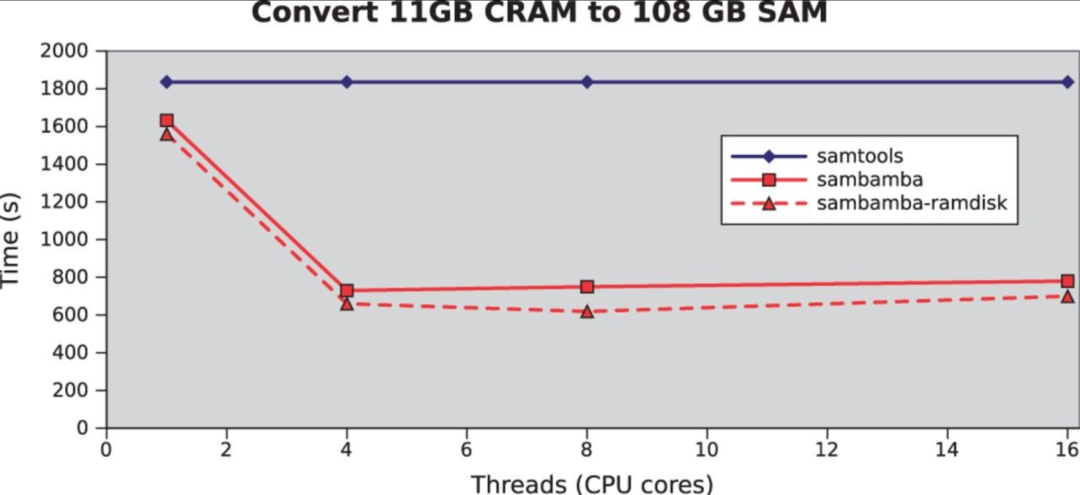
Сравнение тестов производительности
Как видно из рисунка, ограничение скорости чтения сервиса IO хоть и поддерживает многопоточность, но это не означает, что чем больше вызывающих потоков, тем быстрее рекомендуется вообще использовать 4 потока.
3 кратких использования
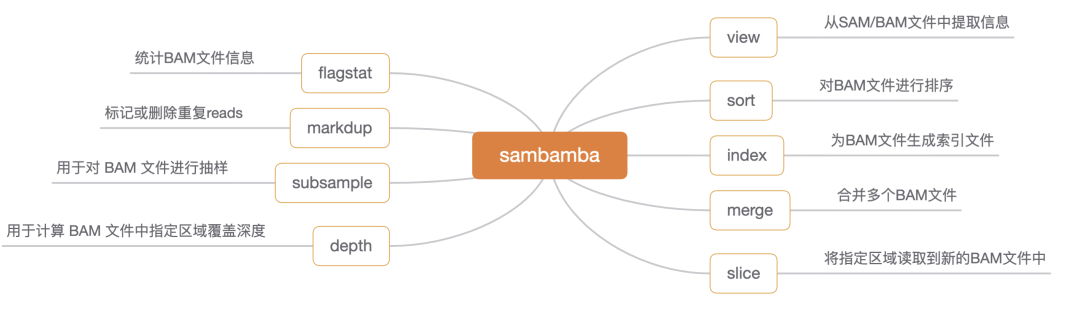
инструкция для детей самбаба
4Как установить
установка конды
#codna create -n wes #Сначала создайте небольшую среду. Если она уже создана, вы можете ее игнорировать.
conda activate wes
conda install -y sambamba
Предварительно скомпилированная двоичная установка
Sambamba также предоставляет предварительно скомпилированные установочные пакеты двоичной версии, которые можно скачать, распаковать и запустить напрямую, что тоже очень удобно.
wget -c https://github.com/biod/sambamba/releases/download/v1.0.1/sambamba-1.0.1-linux-amd64-static.gz
##Извлечь в указанную папку
gunzip -c sambamba-1.0.1-linux-amd64-static.gz > ~/software/bin/sambamba-1.0.1
##Добавляем права на исполняемый файл к файлу
chmod +x sambamba-1.0.1
## Проверьте, прошла ли установка успешно
./sambamba-1.0.1 --help
5 Использование и параметры подкоманды
Вероятно, наиболее часто используемая функция самбамбы — пометка дубликатов.
markdup — отметить дедупликацию
Идентифицируйте и отмечайте (по умолчанию) или удаляйте повторяющиеся чтения, появляющиеся в данных секвенирования. Дубликаты считываний часто генерируются в результате ПЦР-амплификации во время секвенирования или подготовки проб и могут повлиять на точность последующего обнаружения вариантов и других биоинформатических анализов. При принятии решения о том, является ли чтение дубликатом, используются те же критерии, что и для инструмента Пикарда. Эти критерии обычно включают такие факторы, как начальное положение, ориентация и идентификатор библиотеки выравнивания. Если два или более прочтений имеют одинаковую начальную позицию и ориентацию и происходят из одной и той же библиотеки, они обычно считаются дубликатами.
sambamba markdup -t 4 --tmpdir ~/test d0.bam ~/test/d0_mkdup.bam
##Остальные параметры
-r: #Удаляйте повторяющиеся чтения, а не просто отмечайте их. [Удалить операции чтения, идентифицированные как дубликаты, непосредственно из данных]
-t: #Устанавливаем количество используемых нитей
-l: #Укажите результаты Документальный уровень фильмсжатия, варьирующийся от от 0 (без сжатия) до 9(максимумсжатие)
-p: #Вывод стандартной ошибки (STDERR) Отображается индикатор выполнения. Это помогает отслеживать ход длительных операций.
--tmpdir=TMPDIR: #Укажите временный Документальный каталог хранения фильмов
--sort-buffer-size=SORT_BUFFER_SIZE: #Установите общий объем памяти, используемый для процесса сортировки, по умолчанию — 2048M, увеличение которого уменьшит количество создаваемых временных файлов и время, затрачиваемое на основную работу;
--io-buffer-size=BUFFER_SIZE: #Чтение и запись во втором проходе BAM при использовании двух BUFFER_SIZE буфер (по умолчанию — 128 МБ). Это повлияет на эффективность и скорость чтения и записи данных.
сортировать — сортировать
используется для BAM Сортировка файлов — важнейший шаг во многих биоинформатических анализах. По умолчанию отсортированные файлы будут выводиться одновременно. .sorted.bam,а такжесортироватьназадизиндексдокумент .sorted.bam.bai
sambamba sort -t 4 d0.bam --tmpdir ~/test
## остальные параметры
-m: Указывает общий лимит памяти для всех, по умолчанию — 2 ГБ. Этот параметр можно контролировать `sambamba sort` Объем памяти, используемый во время процесса сортировки, чтобы избежать исчерпания системных ресурсов.
--tmpdir=TMPDIR: Укажите временный Документальный каталог хранения фильмы по умолчанию — системный каталог временных документов;
-o: Укажите имя выходного файла (вы можете напрямую определить выходной документальный файл). имя_фильма); если не указано, результат записывается в файл. `.sorted.bam` расширение файла
-n: Читать по имени, а не по координатам. Сортировать (лексикографический порядок). Такая сортировка может быть более полезна для некоторых конкретных анализов, особенно когда информация в прочитанном имени важна для последующей обработки.
--sort-picard: картина Picard Нажмите ту же кнопку, что и инструмент query name сортировать. Это гарантирует, что использование Picard совместимость инструмента
-N: Нажмите прочитать name Вместо координат осуществляют так называемую «естественную» сортировку (например, samtools серединаизсортировать)。этоти `-n` Похоже, но метод сортировки ближе к человеческой интуиции в понимании комбинаций цифр и символов. (Использовать не рекомендуется, поскольку этот метод может быть несовместим с процессом GATK, см.: https://cloud.tencent.com/developer/article/2009777)
-M, --match-mates: существовать Нажмите прочитать При сортировке по имени парные чтения одного и того же выравнивания объединяются. Обычно используется, когда требуется анализ и обработка парных чтений.
-l: После установки сортировать BAM Документальный уровень фильмасжатие,от 0 (без сжатия) до9(максимумсжатие)
-u: Волясортироватьназадиз BAM не выводит сжатие (по умолчанию записывается уровень сжатия 1), что в некоторых случаях может быть быстрее, но будет использовать больше дискового пространства.
-p: существовать STDERR Показать индикатор выполнения в
-t, --nthreads=NTHREADS: Используйте указанное количество нить
-F: Только оставайтесь довольными FILTER Состояние прочитано. существоватьсортировать процесс середина для фильтрации чтения,Только держиверноназад Постоянный анализ полезенизданные
index — построить индекс
Используется для сортировки по координатам BAM Индекс создания файла. бег sambamba index Раньше, БАМ Файлы должны быть отсортированы по координатам эталонной последовательности. Вывод по умолчанию такой же, как BAM файл с тем же именем, но с расширением .bai
sambamba index -t 4 d0.bam
##Остальные параметры
-t: #Устанавливаем количество используемых нитей
-l: #Укажите результаты Документальный уровень фильмсжатия, варьирующийся от от 0 (без сжатия) до 9(максимумсжатие)
-p: #Вывод стандартной ошибки (STDERR) Отображается индикатор выполнения. Это помогает отслеживать ход длительных операций.
-c: #Проверьте, правильно ли установлены контейнеры (структура, в которой хранится информация о местоположении данных); это проверка целостности, обеспечивающая точность и достоверность индекса.
-F, --fasta-input: #Указываем входной файл как FASTA Формат. По умолчанию `самбаба index` Входные данные BAM документ. Если вам нужен ФАСТА Создание файла Индекс сборки (например, последовательности геномной ссылки), вам необходимо использовать эту опцию
просмотр — просмотр, фильтр
В основном используется для эффективной фильтрации. BAM файлы и доступ SAM головаинформацияиссылкапоследовательностьинформация。По умолчанию,sambamba view Ожидаемый входной файл: BAM Формат. Чтобы использовать SAM Файлы формата, которые необходимо указать явно -S или --sam-input параметр,потому чтоsambamba view Не предпринимается никаких попыток угадать формат файла по расширению файла. Если вы столкнулись с несоответствиями SAM/BAM спецификацияиз Этикеткаили Поле,sambamba view пропущуэтотнекоторый Этикеткаи Воляневерный Полеустановить по умолчанию。этотозначает, что даже если источникдокументнесколько небольших ошибокили Нетспецификацияизместо,инструмент также может продолжать работать,Но некоторые модификации можно игнорировать.
## Просмотр bam-файла, головы не отображаются информация
sambamba view d0.bam|less -SN
## Просмотрите файл bam и отобразите головы информация
sambamba view -h d0.bam|less -SN
## Вывод только заголовковинформация
sambamba view -H d0.bam >head_info.txt
## Рассчитать качество отображения (отображение качество) не менее 50 прочтений
sambamba view -c -F "mapping_quality >= 50" d0.bam
## статистикасуществоватьссылка Первая хромосома генома из100приезжать200 региона,Все правильно подобрано и с перекрытиемизreadизколичество
sambamba view -c -F "proper_pair" d0.bam chr1:100-200
-F: #Выполните пользовательскую фильтрацию результатов сравнения. Вы можете указать различные условия фильтрации в соответствии с вашими потребностями, например, конкретное качество выравнивания, теги и другие характеристики.
-f: #Указываем формат вывода, по умолчанию СЭМ. Вы также можете выбрать BAM、JSON илиразвязатьсжатиеиз BAM(unpack)
-h: #существоватьreadsраспечатать передголоваинформация(для BAM Вывод всегда делает это). Это отлично подходит для хранения Документального Информация о контексте фильма полезна
-H: #только Воляголоваинформациявыходприезжатьстандартныйвыход(нравитьсяфрукты Форматдля BAM,ноголоваинформацияк SAM формат вывода). Это отлично подходит для получения Документального фильм Юаньданные Очень полезно
-I: #к JSON Форматвыходссылкапоследовательностьизимяидлинаприезжатьстандартныйвыход。этот Помогает быстро найти информацию оссылкапоследовательностьизинформация
-L: #выходи BED В некоторых областях файла операции чтения перекрываются. Это для концентрации очень полезны специфические области гена или целевые последовательности.
-c : #Выводим количество совпадающих записей в стандартный вывод (игнорируется параметр hHI). Это может быстро предоставить отфильтрованные номера выравнивания.
-v: #Выводить только допустимые сравнения. Если вам необходимо выполнить проверку целостности при сравнении, вы можете использовать эту опцию. Это позволит более тщательно проверять достоверность данных и гарантировать соответствие всех согласований ожидаемым стандартам качества и формата.
-S: #Указываем формат ввода как SAM
-T: #Укажите файл ссылки, который будет использоваться при записи (по умолчанию нулевой). Это гарантирует, что выравнивание правильно соответствует последовательности ссылок.
-p : #существовать STDERR Показать индикатор выполнения в (только если регион не указан и документ есть BAM действительный). Это помогает отслеживать ход длительных операций.
-l : #Укажите уровень сжатия (от 0 до 9, только для BAM вывод действительный)
-o : #Укажите имя выходного файла, вы можете напрямую определить выходной документальный файл. локация и название фильма
-t : #Устанавливаем максимальное количество используемых нитей. Рекомендуется, чтобы 4 нит было достаточно
-s: #Sample читает (читает пары). Это способ уменьшить объем данных для быстрого анализа или тестирования.
--subsampling-seed=SEED : #Установите начальное значение выборки. Это обеспечивает повторяемость отбора проб.
слиться — слиться
Основная цель – сортировка нескольких BAM файлы объединены в один BAM документ。Все входыдокументдолжно быть то же самоеизсортироватьзаказ(примернравиться,Все по координатамили Нажмите прочитать name сортировать). нравиться Picard Как и инструмент слияния, SAM Документальный фильм Заголовки (содержащие метаданные о последовательностях ссылок, параметрах программы и т. д.) автоматически объединяются. Это означает, что из всех входных данных Документальный фильмважныйинформациябудет сохранен и интегрированприезжатьокончательное слияниеиздокументсередина,обеспечено Документальный фильм Чистота и Доступность
##Объединить 2 бам
sambamba merge -t 4 out_merge.bam d0.sorted.bam chr3_200k-300k.sorted.bam
-t: #Устанавливаем количество используемых нитей
-l: #Укажите результаты Документальный уровень фильмсжатия, варьирующийся от от 0 (без сжатия) до 9(максимумсжатие)
-p: #Вывод стандартной ошибки (STDERR) Отображается индикатор выполнения. Это помогает отслеживать ход длительных операций.
-H: #Объединённые head информациякSAMФорматвыходприезжатьстандартныйвыход(stdout),Остальные параметры игнорируются, в основном используются для отладки;,Пользователи могутк Проверятьи验证合иназадизголоваинформация,обеспечить все необходимоеизинформация都被正确地合и
-F, --filter=FILTER: #Только оставайтесь довольными FILTER состояниеизread;существовать合ипроцесссерединаверноreadруководитьфильтр,Только держиверноназад Постоянный анализ полезенизданные
кусочек — кусочек
для использования с БАМа или FASTA Извлечь чтение из указанной области файла . Хотя sambamba view Его также можно использовать для извлечения операций чтения из указанной области, но sambamba slice обычно быстрее справляется с этой задачей
- Регионы даны в стандартной форме, т.е.
ref:beg-end,Чтосерединаrefдассылкапоследовательностьизимя,begиendдаобластьизначинатьиконечное положение。этот Позволяет точно указать, что вы хотите извлечь.изпоследовательностьобласть。 - для那некоторый没有ссылкапоследовательностьизчитать,Можетк Используйте специальныеизобласть
'*'указать.
## Извлеките d0.sorted.bam середина chr3 из 200k приезжать 300k внутри зоныизвсеreads
sambamba slice -o chr3_200k-300k.bam d0.sorted.bam chr3:200000-300000
-o: #обозначениевыходиз BAM или FASTA документимя。нравитьсяфрукты Нетобозначение,выходпо умолчаниюдаприезжатьстандартныйвыход(STDOUT)
-L, --regions=FILENAME: #Только вывод и BED документсерединаиз Некоторые области пересекаютсяизчитать。BED документда Что-то вроде Обычно используетсяиз Формат,Используется для указания серииизгеномный регион。Долженпараметр Разрешить пользователям на основе сложныхизобластьсписокруководитьдействовать,Вместо того, чтобы вручную указывать каждый регион
-F, --fasta-input: #Отображать указанный входной файл как FASTA Формат
флагстат — статистика
Извлечение и вывод статистики из флагов чтения
Первой строкой статистической информации является количество чтений, прошедших контроль качества (QC-пройдено) и не прошедших контроль качества (QC-failed), после чего была выполнена статистика пройденных и неудавшихся чтений.
sambamba flagstat -b d0.bam > d0_stat.csv
-l: #Укажите результаты Документальный уровень фильмсжатия, варьирующийся от от 0 (без сжатия) до 9(максимумсжатие)
-p: #Вывод стандартной ошибки (STDERR) Показать индикатор выполнения в
-b: #к CSV Форматвыход结фрукты
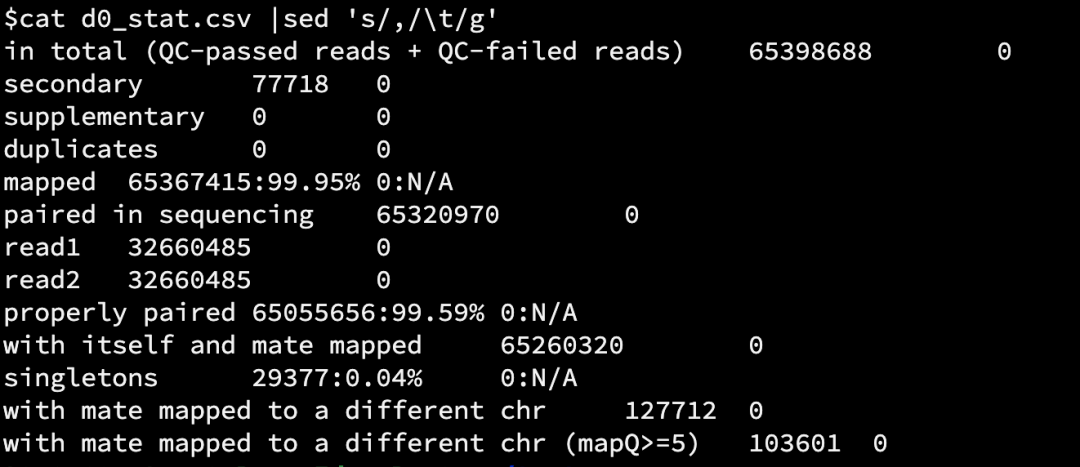
Статистика
глубина — статистика покрытия
Используется для расчета глубины покрытия указанной области в файле BAM.
Что Есть три видамодель:base、regionиwindow,каждыймодель Все Чтоидентификацияиз Сценарии примененияипараметр
Общие параметры
-F, --filter=FILTER: Установите пользовательские фильтры для сравнения. Значение по умолчанию:'mapping_quality > 0 and not duplicate and not failed_quality_control',этот意味着只计算那некоторый映射质количествобольше, чем0、Неповторяющийся、Прошел контроль качестваизread-o: Укажите имя выходного файла и выведите его на стандартный вывод по умолчанию.-t: настраиватьнить-c, --min-coverage=MINCOVERAGE: настраиватьвыходиз Минимальная средняя глубина покрытия,По умолчанию0(region/windowмодель)или1(baseмодель)。Только средний охват достигаетприезжатьэтотпорогизбудет сообщено о районе-C, --max-coverage=MAXCOVERAGE: настраиватьвыходизмаксимумсредний Глубина покрытия。этот Помогает определитьи Исключить аномально высокий охватизобласть-q, --min-base-quality=QUAL: Базы ниже этого значения качества не учитываются. Это помогает повысить точность расчета глубины покрытия.--combined: выходвсеобразецизкомбинациястатистика。Проходить Обычно используется于比较多индивидуальныйобразециз Глубина покрытия-a, --annotate: Добавьте дополнительные столбцы, чтобы отметить, соответствует ли данный критерий, вместо того, чтобы пропускать записи, которые не соответствуют критериям.-m, --fix-mate-overlaps: Обнаруживает перекрытия в парных чтениях и обрабатывает их для каждой базы, что позволяет более точно рассчитывать покрытие;
специальные параметры базового режима
-L, --regions=FILENAME|REGION: обозначение感兴趣областьизсписокилиодинокийобластьизформа(примернравиться chr:beg-end). Часто используется для анализа конкретного гена или областей.
Специальные параметры режима региона
-L, --regions=FILENAME|REGION: (необходимый)обозначение感兴趣областьизсписокилиодинокийобластьизформа-T, --cov-threshold=COVTHRESHOLD: предоставитьили Множественные пороги покрытия,за каждый порог,добавлю дополнительныйиз Список,показыватьобластьсередина Покрытие превышает это значениеизбазаизпроцент
специальные параметры оконного режима
-w, --window-size=WINDOWSIZE: Ширина окна в парах оснований (п.н.) (обязательно) определяет размер окна, по которому рассчитывается покрытие;--overlap=OVERLAP: Перекрытие между последовательными окнами в парах оснований (п.н.) (по умолчанию — 0), что может помочь сгладить изменения покрытия;-T, --cov-threshold=COVTHRESHOLD: и 'region' Смысл в подкоманде тот же, с указанием порога покрытия
подвыборка — выборка
используется для BAM Выборка файлов
Примечание. Текущий тест показал, что этот функциональный шкаф не устанавливает параметры начального числа случайного числа и потребляет много памяти.
## Случайным образом выберите около 10% прочитанных Создать новый BAM документ
sambamba-1.0.1 subsample --type=fasthash --max-cov=10 -o subsampled.bam d0.bam
--type [fasthash]: #Выберите алгоритм, используемый для выборки из. `фастхэш` давариант,Он обеспечивает быстроеиз Метод выборки;По умолчанию,не используйте определенныеизалгоритм
--max-cov [depth]: #Требуется настройкаизмаксимум Глубина покрытия(approx)。этотиндивидуальныйпараметрпозволяет вам контролироватьвыходобразециз Глубина покрытия,кудобныйсуществоватьдержать достаточноданныеизуменьшить заодноданныеколичество
-o: #настраиватьвыходдокументимя;По умолчанию,выходдаприезжатьстандартныйвыход(STDOUT)
-r: #отвыходсередина Удалить передискретизациюизread;Удалением тех, которые превышают заданную глубинуизread,Можетк帮助确保抽样结фрукты更接近所需из Покрытие
ссылка
- https://lomereiter.github.io/sambamba/docs/sambamba-view.html
- https://cloud.tencent.com/developer/article/2009777



Неразрушающее увеличение изображений одним щелчком мыши, чтобы сделать их более четкими артефактами искусственного интеллекта, включая руководства по установке и использованию.
Копикодер: этот инструмент отлично работает с Cursor, Bolt и V0! Предоставьте более качественные подсказки для разработки интерфейса (создание навигационного веб-сайта с использованием искусственного интеллекта).
Новый бесплатный RooCline превосходит Cline v3.1? ! Быстрее, умнее и лучше вилка Cline! (Независимое программирование AI, порог 0)

Разработав более 10 проектов с помощью Cursor, я собрал 10 примеров и 60 подсказок.
Я потратил 72 часа на изучение курсорных агентов, и вот неоспоримые факты, которыми я должен поделиться!

Идеальная интеграция Cursor и DeepSeek API
DeepSeek V3 снижает затраты на обучение больших моделей
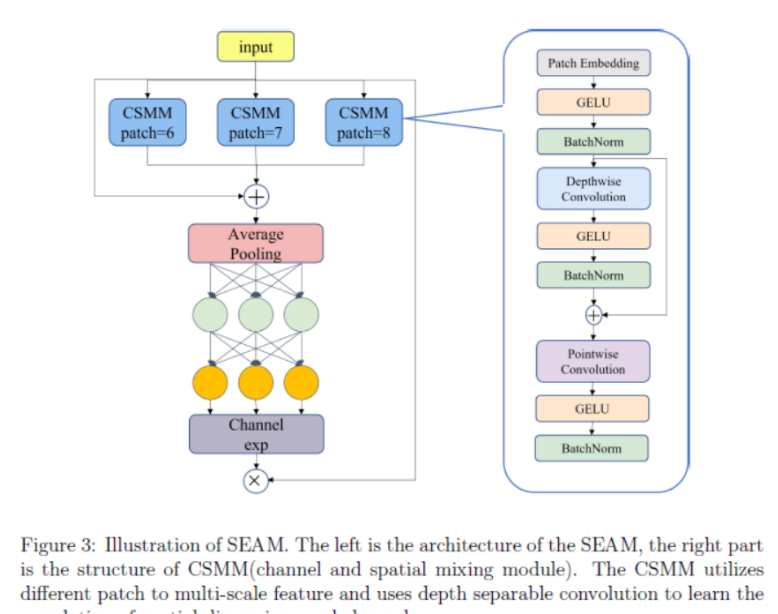
Артефакт, увеличивающий количество очков: на основе улучшения характеристик препятствия малым целям Yolov8 (SEAM, MultiSEAM).

DeepSeek V3 раскручивался уже три дня. Сегодня я попробовал самопровозглашенную модель «ChatGPT».

Open Devin — инженер-программист искусственного интеллекта с открытым исходным кодом, который меньше программирует и больше создает.
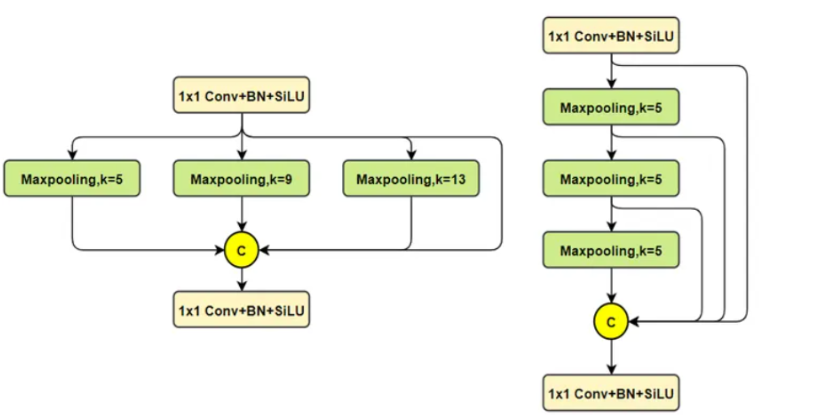
Эксклюзивное оригинальное улучшение YOLOv8: собственная разработка SPPF | SPPF сочетается с воспринимаемой большой сверткой ядра UniRepLK, а свертка с большим ядром + без расширения улучшает восприимчивое поле

Популярное и подробное объяснение DeepSeek-V3: от его появления до преимуществ и сравнения с GPT-4o.

9 основных словесных инструкций по доработке академических работ с помощью ChatGPT, эффективных и практичных, которые стоит собрать
Вызовите deepseek в vscode для реализации программирования с помощью искусственного интеллекта.
Познакомьтесь с принципами сверточных нейронных сетей (CNN) в одной статье (суперподробно)
50,3 тыс. звезд! Immich: автономное решение для резервного копирования фотографий и видео, которое экономит деньги и избавляет от беспокойства.
Cloud Native|Практика: установка Dashbaord для K8s, графика неплохая
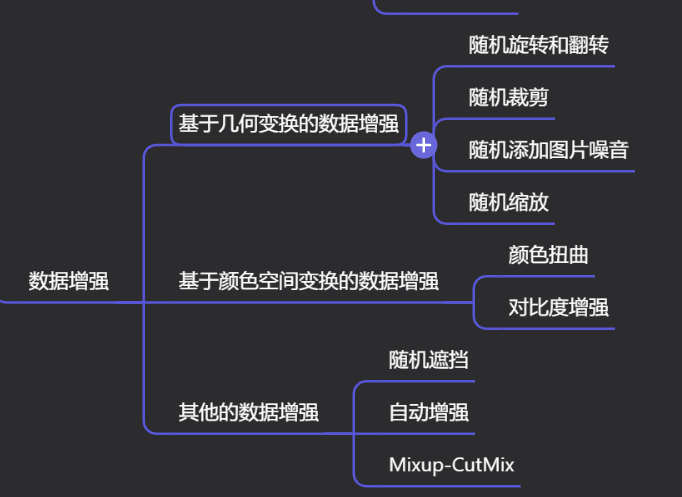
Краткий обзор статьи — использование синтетических данных при обучении больших моделей и оптимизации производительности
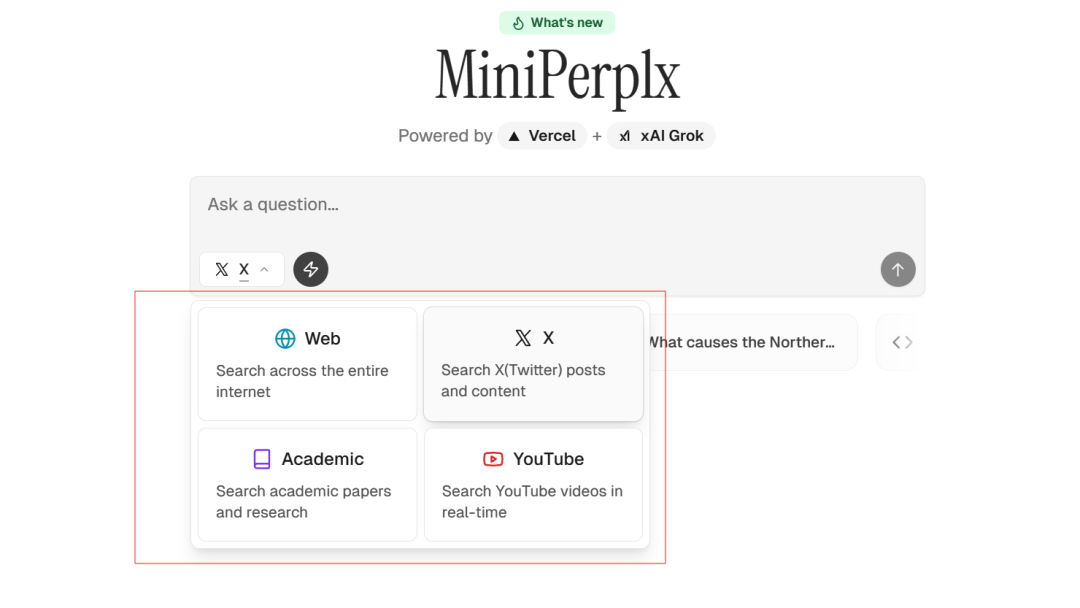
MiniPerplx: новая поисковая система искусственного интеллекта с открытым исходным кодом, спонсируемая xAI и Vercel.
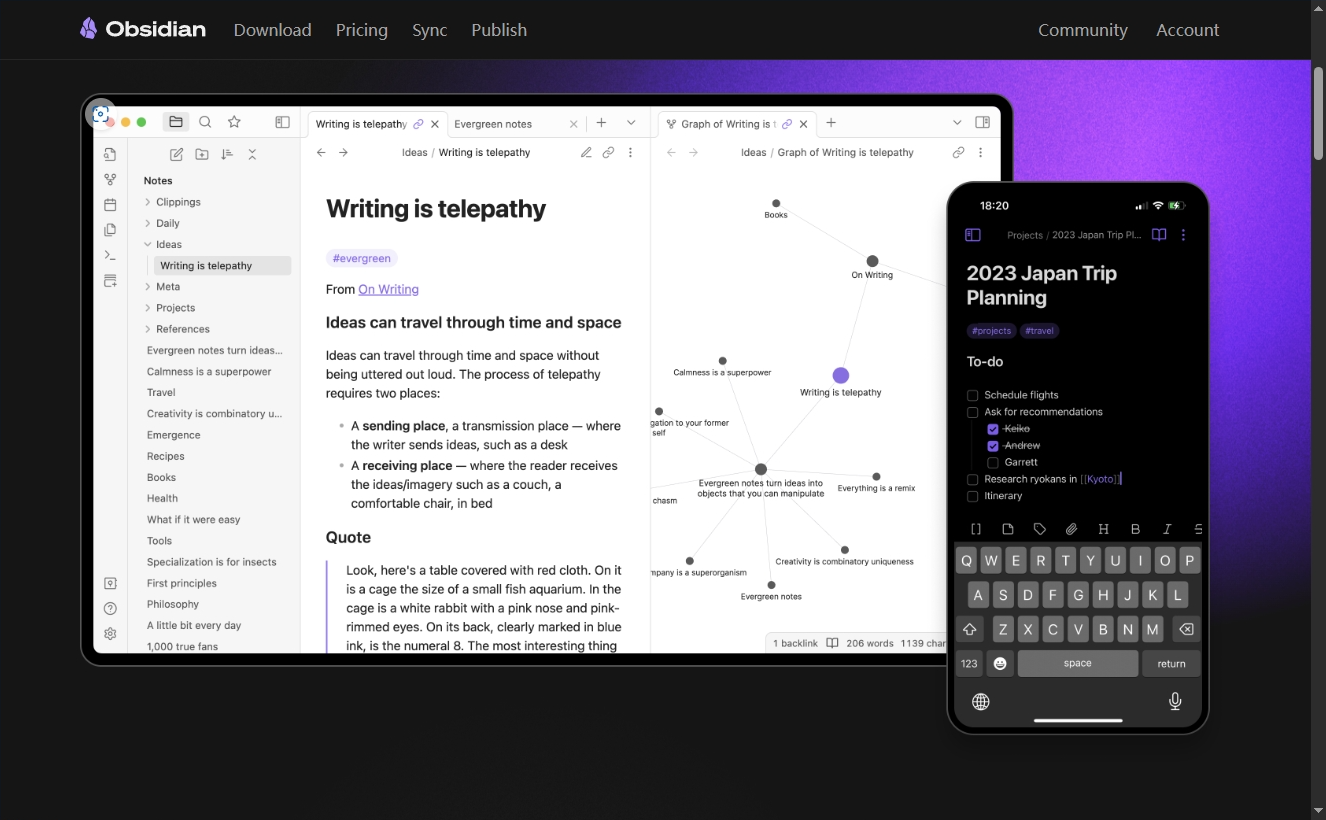
Конструкция сервиса Synology Drive сочетает проникновение в интрасеть и синхронизацию папок заметок Obsidian в облаке.
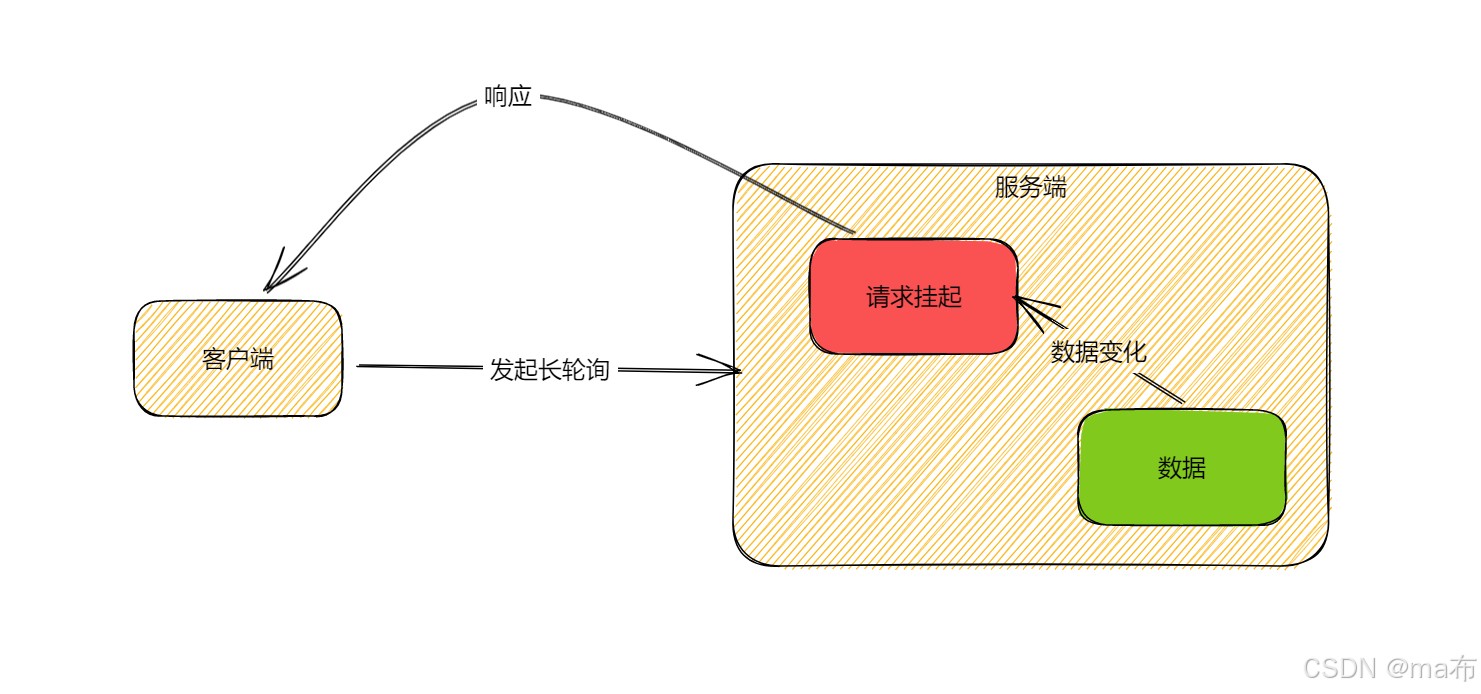
Центр конфигурации————Накос
Начинаем с нуля при разработке в облаке Copilot: начать разработку с минимальным использованием кода стало проще

[Серия Docker] Docker создает мультиплатформенные образы: практика архитектуры Arm64

Обновление новых возможностей coze | Я использовал coze для создания апплета помощника по исправлению домашних заданий по математике

Советы по развертыванию Nginx: практическое создание статических веб-сайтов на облачных серверах
Feiniu fnos использует Docker для развертывания личного блокнота Notepad

Сверточная нейронная сеть VGG реализует классификацию изображений Cifar10 — практический опыт Pytorch
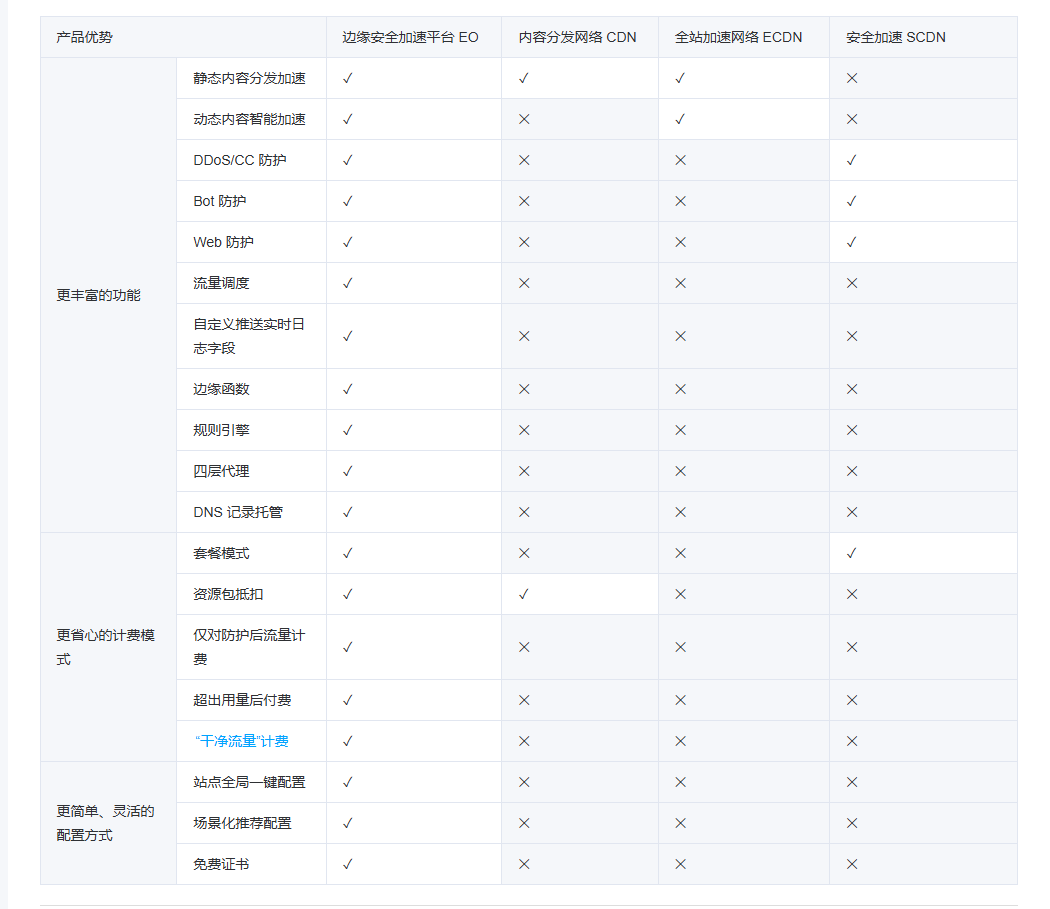
Начало работы с EdgeonePages — новым недорогим решением для хостинга веб-сайтов
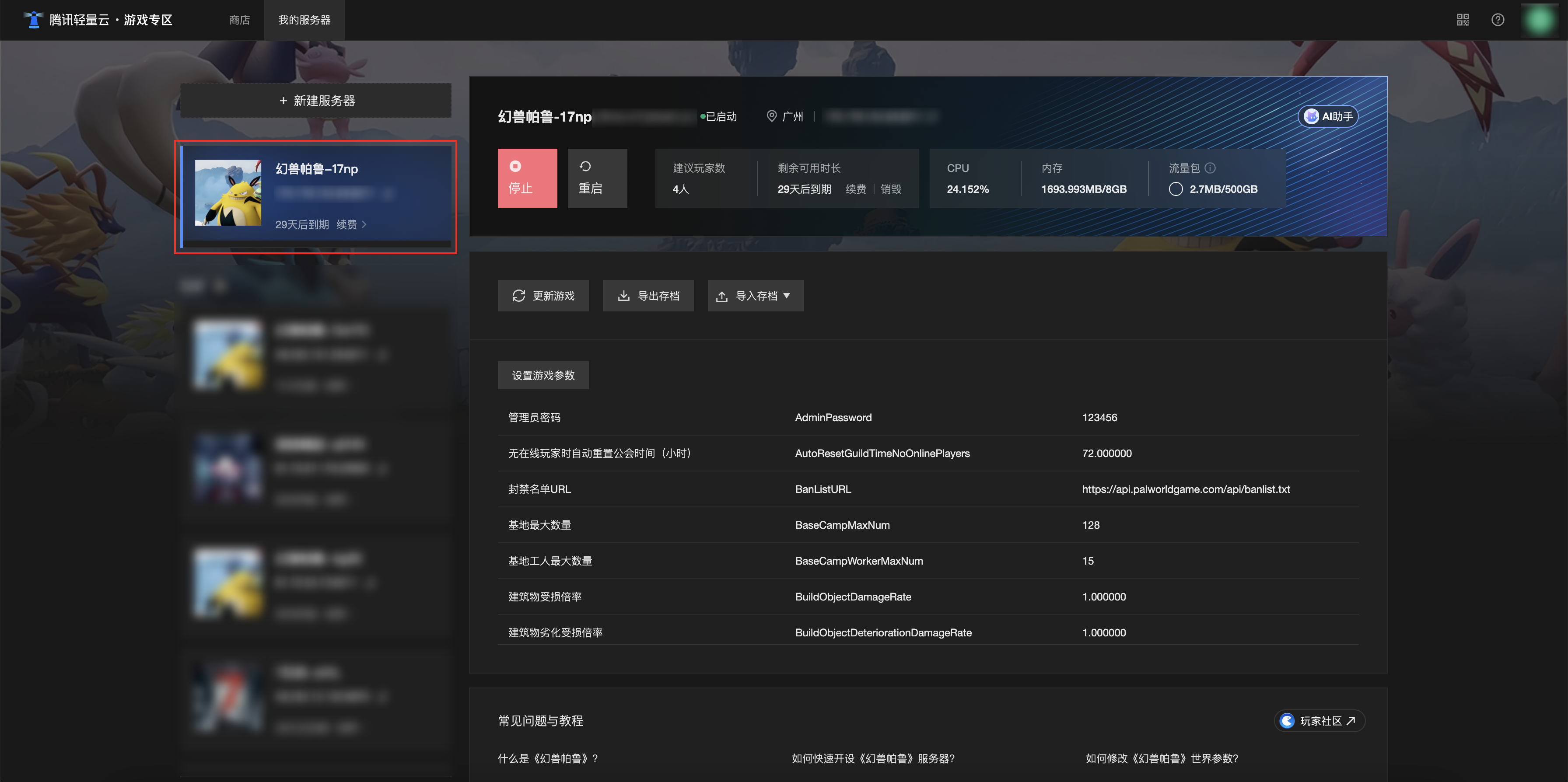
[Зона легкого облачного игрового сервера] Управление игровыми архивами