Лучшее руководство по секвенированию отдельных клеток (16): все, что вы хотите знать о скорости РНК, находится здесь.
Примечание автора В этой главе подробно объясняются три квазипоследовательные модели, основанные на скорости РНК, включая модель стационарного состояния, модель EM и модель глубокого обучения, а также сравниваются применимые сценарии и вычислительные характеристики различных моделей. Это руководство было впервые опубликовано в журнале Single Cell’s Best Chinese Tutorial. Перепечатка без разрешения запрещена. Общее количество слов|Расчетное время чтения: 5000|10 минут. ——Звездная ночь
5.2 Скорость РНК
1. Предыстория
Сбор данных по одной клетке позволяет изучать биологические процессы, такие как раннее развитие, с высоким разрешением. Хотя после секвенирования клетки,оно уничтожено,Поэтому его определяющие свойства не могут быть измерены снова в более поздний момент времени.,Но мы можем изучать клетки в разных состояниях с помощью снимков. также,Стоит отметить, что,Традиционные экспериментальные методы не только не позволяют измерить общие свойства клеток в разное время.,и не смог измерить, насколько быстро произошли эти клеточные изменения.。Но мы можем пройтивывод траектории(TI)Инструменты предметной области для восстановления пространства состояний ячейки。Однако традиционные алгоритмы измеряют его только по двум измерениям: сходству клеток и матрице экспрессии.,Соответствующий контент в некоторой степени отсутствует.
2. Скорость РНК
Изменения в профиле транскриптома клетки запускаются рядом событий: в широком смысле ДНК транскрибируется с образованием так называемой несплайсированной информационной РНК-предшественника (пре-м РНК). Несплайсированная пре-м РНК содержит области, связанные с трансляцией (экзоны), а также некодирующие области (интроны). Эти некодирующие области подвергаются сплайсингу, то есть удаляются, в результате чего образуется сплайсированная зрелая м РНК. Хотя протоколы секвенирования одноклеточной РНК (scRNA-seq) не способны захватывать транскриптом в несколько моментов времени, они содержат информацию, необходимую для различения несплайсированных и сплайсированных считываний м РНК.
Идентификация несрезанных и сдвинутых чтений позволяет строить динамические модели, описывающие динамику сдвига, и выводить соответствующие веса модели на основе данных об одной ячейке. Изменения в сплайсированной РНК, описываемые моделью, называются скоростью РНК. Современные модели скорости РНК предполагают, что модель, специфичная для гена,
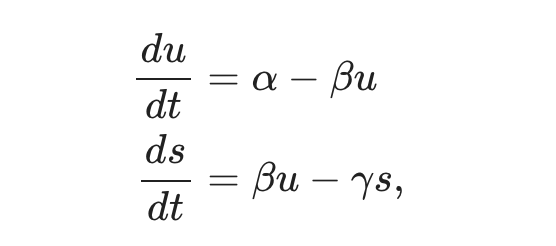
в,αэто скорость транскрипции,β это скорость сдвига,γ – скорость деградации расщепленной РНК. Хотя динамика каждого гена моделируется независимо,Для простоты обозначений,Мы собираемся избавиться от индексаg . Хотя область оценки параметров в динамических системах интенсивно изучается, алгоритмы вывода требуют знания времени, связанного с каждым наблюдением. Следовательно, эти традиционные методы не могут быть применены для определения скорости РНК и параметров ее модели в контексте данных scRNA-seq.
3. Модельная гипотеза
Секвенирование отдельных клеток представляет собой моментальный снимок данных,Поэтому его нельзя построить в зависимости от времени. Напротив,традиционныйRNAСкоростные методы основаны на изучении специфичных для клеток кортежей каждого гена.(u, s),вuиs分别代表未резатьирезатьизRNA。Совокупность этих кортежей образует так называемую фазовую карту.。гипотетическая транскрипция、Скорость сдвига и деградации постоянна.,Фазовая диаграмма приобретает миндалевидную форму. Верхняя дуга соответствует фазе индукции,Нижняя дуга соответствует фазе торможения. Однако,Поскольку данные реального мира зашумлены,Построение графика количества неразрезанных и разрезанных элементов не восстанавливает ожидаемую миндалевидную форму. Напротив,данные необходимо сначала сгладить. Классически,Этот этап предварительной обработки включает в себя усреднение экспрессии генов каждой клетки на графике сходства клеток.
В этом уроке мы представим стационарную модель, модель EM и модель глубокого обучения соответственно.
from tqdm import tqdm
from multiprocessing import Lock
tqdm.set_lock(Lock()) # manually set internal lock
tqdm.write("test")
import scanpy as sc
import matplotlib.pyplot as plt
import numpy as np
import pandas as pd
import omicverse as ov
ov.plot_set()
# ____ _ _ __
# / __ \____ ___ (_)___| | / /__ _____________
# / / / / __ `__ \/ / ___/ | / / _ \/ ___/ ___/ _ \
# / /_/ / / / / / / / /__ | |/ / __/ / (__ ) __/
# \____/_/ /_/ /_/_/\___/ |___/\___/_/ /____/\___/
# Version: 1.5.9, Tutorials: https://omicverse.readthedocs.io/
4. Загрузите данные
Чтобы продемонстрировать метод в этом уроке, мы будем использовать набор данных scRNA-seq развития поджелудочной железы мышей на эмбриональной стадии (E) 15.5.
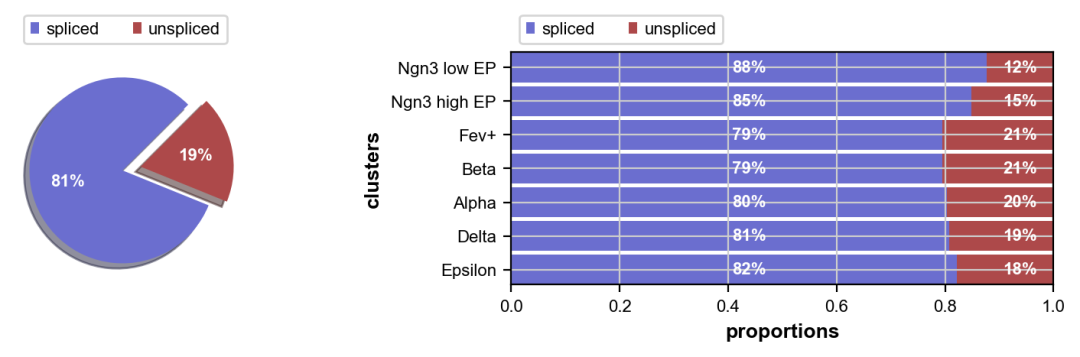
Мы наблюдаем соотношение сплайсированных/несплайсированных чтений, необходимых для оценки скорости РНК;
import cellrank as cr
import scvelo as scv
adata = cr.datasets.pancreas()
scv.pl.proportions(adata)
adata
# AnnData object with n_obs × n_vars = 2531 × 27998
# obs: 'day', 'proliferation', 'G2M_score', 'S_score', 'phase', 'clusters_coarse', 'clusters', 'clusters_fine', 'louvain_Alpha', 'louvain_Beta', 'palantir_pseudotime'
# var: 'highly_variable_genes'
# uns: 'clusters_colors', 'clusters_fine_colors', 'day_colors', 'louvain_Alpha_colors', 'louvain_Beta_colors', 'neighbors', 'pca'
# obsm: 'X_pca', 'X_umap'
# layers: 'spliced', 'unspliced'
# obsp: 'connectivities', 'distances'
5. Предварительная обработка данных
Гены без достаточного количества сплайсированных/несплайсированных были отсеяны, нормализованы и зарегистрированы данные конверсии, ограничивающиеся высоковариабельными верхними генами. Далее вычисляются главные компоненты и моменты оценки скорости.
scv.pp.filter_and_normalize(
adata, min_shared_counts=20, n_top_genes=2000, subset_highly_variable=False
)
sc.tl.pca(adata)
sc.pp.neighbors(adata, n_pcs=30, n_neighbors=30, random_state=0)
scv.pp.moments(adata, n_pcs=None, n_neighbors=None)
# Filtered out 22024 genes that are detected 20 counts (shared).
# Normalized count data: X, spliced, unspliced.
# Extracted 2000 highly variable genes.
# Logarithmized X.
# computing PCA
# on highly variable genes
# with n_comps=50
# log1p(adata)
# finished (0:00:00)
# computing neighbors
# using 'X_pca' with n_pcs = 30
# finished: added to `.uns['neighbors']`
# `.obsp['distances']`, distances for each pair of neighbors
# `.obsp['connectivities']`, weighted adjacency matrix (0:00:04)
# computing moments based on connectivities
# finished (0:00:00) --> added
# 'Ms' and 'Mu', moments of un/spliced abundances (adata.layers)
6. Стационарная модель.
Мы оцениваем скорость РНК, предполагая, что гены независимы.,А основная динамика задается вышеупомянутой Моделью. также,Мы также предполагаем, что (1) динамика достигает равновесия,(2) Скорость постоянна,а также(3)Все гены имеют один、обычная скорость сдвига。Итак, мы сделаем это Модельназываетсястационарная модель。Само установившееся состояние расположено в правом верхнем углу фазовой диаграммы.(индукционная фаза)и其原点(фаза торможения)。На основе этих крайних квантилей,стационарная модельИспользуйте линейную регрессию, чтобы оценить коэффициенты устойчивого состояния.。Затем,Скорость РНК определяется как остаток от этой подгонки.
хотястационарная модельНаправление развития может быть успешно восстановлено в некоторых системах.,Но он по своей сути ограничен из-за предположений Модели. Два предположения, которые легко нарушить, заключаются в том, что все гены имеют одинаковую скорость сдвига.,и состояние равновесия, наблюдаемое в ходе эксперимента. поэтому,Экстраполяция в таких обстоятельствах даст ошибочные результаты. также,стационарная модельтолько рассмотретьданныеподмножество,и только вывести установившееся соотношение,Вместо каждого параметра Модели.
Мы на стационарной модель РассчитатьRNAскорость。в этом случае,мы используемscVeloизvelocityфункция,и установитьmode="deterministic"。
scv.tl.velocity(adata, mode="deterministic")
# computing velocities
# finished (0:00:00) --> added
# 'velocity', velocity vectors for each individual cell (adata.layers)
Хотя мы не одобряем чрезмерную интерпретацию проецирования многомерного вектора скорости на низкоразмерное представление данных,ноscVelo提供了一个简单из方法来实现这一点。
scv.tl.velocity_graph(adata, n_jobs=8)
scv.pl.velocity_embedding_stream(adata, basis="umap", color="clusters")
# computing velocity graph (using 8/12 cores)
# 0%| | 0/2531 [00:00<?, ?cells/s]
# finished (0:00:08) --> added
# 'velocity_graph', sparse matrix with cosine correlations (adata.uns)
# computing velocity embedding
# finished (0:00:00) --> added
# 'velocity_umap', embedded velocity vectors (adata.obsm)
7. ЭМ-модель
чтобы преодолетьстационарная модельиз局限性,Было предложено несколько методов расширения.。迄今为止最受欢迎из是在scVelo中实施изЭМ-модель 。ЭМ-модельБольше не предполагается, что установившееся состояние достигнуто.,Также не предполагается, что гены имеют общую скорость сдвига. также,Все точки данных используются для вывода полного набора параметров.,А также потенциальное время сдвига, специфичное для генов и клеток. Модель. Алгоритм использует структуру максимизации ожидания (EM) для оценки параметров. Ненаблюдаемые переменные, обнаруженные на этапе E, включают время и состояние (индуцированное, тормозящее или устойчивое состояние) каждой клетки. Все остальные параметры модели выводятся на этапе M.
ХотяЭМ-модельБольше не зависит отстационарная модельиз关键假设,Поэтому он имеет более широкое применение,Но предполагаемая скорость РНК все еще может противоречить предыдущим биологическим знаниям. Есть две основные причины этой неудачи: с одной стороны,,ЭМ-модель仍然假设速率是恒定из。поэтому,Всякий раз, когда эти предположения не выполняются,Например, во время созревания эритроцитов,Вывод просто неверен. с другой стороны,Предлагаемая модель, как и ее предшественница, опирается на фазовые диаграммы. поэтому,Когда фазовая диаграмма гена не соответствует ожидаемой форме,Алгоритм по своей сути неуместен и не работает.
чтобы использоватьЭМ-модельвычислитьRNAскорость,首先需要推断резать动力学из参数。выведено изscVeloизrecover_dynamicsфункция处理。
scv.tl.recover_dynamics(adata, n_jobs=8)
# recovering dynamics (using 8/12 cores)
# 0%| | 0/783 [00:00<?, ?gene/s]
# finished (0:00:55) --> added
# 'fit_pars', fitted parameters for splicing dynamics (adata.var)
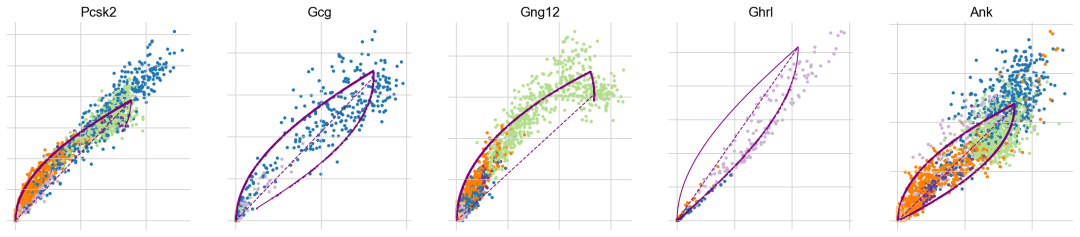
резать Модельиз参数是通过最大化给定из似然функция来推断из。Чтобы изучить, какие геныscVeloПодходит максимально уверенно,Мы можем изучить соответствующую фазовую диаграмму вместе с предполагаемой траекторией (показана фиолетовым цветом) и установившимся коэффициентом (пунктирная фиолетовая линия). здесь,五个显示из基因中有两个呈现出(Pcsk2,Gcg、Ank)杏仁形状из相位图。我们观察приезжать明确из转变.
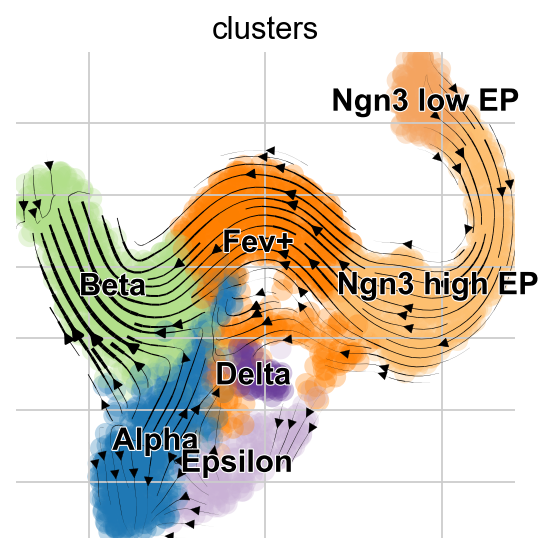
внутри одного типа клеток(Gcg、Ghr1)Или охватывать несколько типов клеток?(Pcsk2,Из Ngn3 low EPприезжатьAlphaиBeta)。И вGng12из情况下,Мы наблюдали популяцию клеток в устойчивом состоянии. Скорее всего, это артефакт из-за недостаточной выборки фенотипического многообразия, окружающего клетки с низким/высоким уровнем EP Ngn3. Хотя ячеек всего несколько из-за небольшого размера кластера. Хотя текущая передовая практика ограничивается ручным анализом соответствия Модели и уверенности в ней.,Но предложенные недавно методы могут помочь автоматизировать этот процесс (новое направление). здесь,GcgиGhrl将被分配一个较低из置信度分数。
top_genes = adata.var["fit_likelihood"].sort_values(ascending=False).index
scv.pl.scatter(adata, basis=top_genes[:5], color="clusters", frameon=False)
После оценки кинетической скорости(Хранить какadata.obsизfit_alpha、fit_beta、fit_gammaСписок)после,Мы можем вычислить скорость,и спроецируйте его в наше встраивание 2D UMAP.
scv.tl.velocity(adata, mode="dynamical")
scv.tl.velocity_graph(adata, n_jobs=8)
scv.pl.velocity_embedding_stream(adata, basis="umap")
# computing velocities
# finished (0:00:01) --> added
# 'velocity', velocity vectors for each individual cell (adata.layers)
# computing velocity graph (using 8/12 cores)
# 0%| | 0/2531 [00:00<?, ?cells/s]
# finished (0:00:08) --> added
# 'velocity_graph', sparse matrix with cosine correlations (adata.uns)
# computing velocity embedding
# finished (0:00:00) --> added
# 'velocity_umap', embedded velocity vectors (adata.obsm)
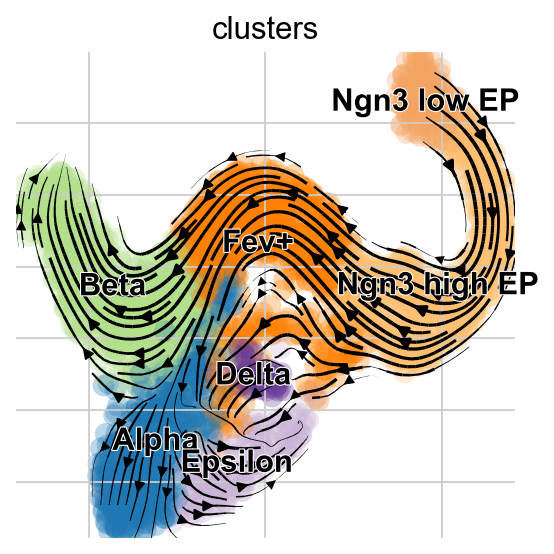
Проекция стационарной модели показывает «рефлюкс» от альфа-клеток к преэндокринным клеткам, но это явление не проявляется в модели EM.
Кинетические модели восстанавливают время основных клеточных процессов. Это основное время представляет собой внутренние часы клетки и приблизительно соответствует фактическому времени, которое клетка проходит во время дифференцировки, основываясь исключительно на ее транскрипционной динамике.
scv.tl.latent_time(adata)
adata.obs['latent_time_EMmodel']=adata.obs['latent_time'].copy()
# computing terminal states
# WARNING: Uncertain or fuzzy root cell identification. Please verify.
# identified 1 region of root cells and 1 region of end points .
# finished (0:00:00) --> added
# 'root_cells', root cells of Markov diffusion process (adata.obs)
# 'end_points', end points of Markov diffusion process (adata.obs)
# computing latent time using root_cells as prior
# finished (0:00:00) --> added
# 'latent_time', shared time (adata.obs)
adata.obs['velocity_pseudotime_EMmodel']=adata.obs['velocity_pseudotime'].copy()
8. Модель глубокого обучения
Хотя эти методы оценки скорости РНК успешно использовались для объяснения динамики отдельных клеток, они также имеют ограничения из-за допущений моделирования и дальнейшего использования. Например, в обоих подходах отсутствует глобальная концепция неопределенности. Следовательно, может быть сложно оценить надежность оценок скорости РНК или решить, насколько хорошо анализ скорости применим к данному набору данных.
veloVI (вариационный вывод скорости) — это глубокая генеративная модель для оценки скорости РНК. VeloVI переформулирует вывод о скорости РНК с помощью модели, которая распределяет информацию между всеми клетками и генами, изучая при этом те же величины, что и в модели EM, а именно кинетические параметры и скрытое время. В этой переформулировке используются преимущества глубокого генеративного моделирования20, которые стали центральными для многих задач омного анализа одиночных ячеек, таких как мультимодальная интеграция данных, моделирование возмущений и коррекция данных. В качестве выходных данных veloVI возвращает эмпирическое апостериорное распределение скорости РНК (матрица клеток-генов-апостериорных образцов), которое можно включить в последующий анализ результатов. Более того, он обеспечивает уровень интерпретации и критики модели, которого не хватало предыдущим подходам, а также значительно повышает гибкость расширений модели.
Мы можем импортировать VeloVI для изучения квазипоследовательных значений в рамках модели глубокого обучения.
#adata = preprocess_data(adata)
from velovi import VELOVI
VELOVI.setup_anndata(adata, spliced_layer="Ms", unspliced_layer="Mu")
vae = VELOVI(adata)
vae.train()
# GPU available: True (cuda), used: True
# TPU available: False, using: 0 TPU cores
# IPU available: False, using: 0 IPUs
# HPU available: False, using: 0 HPUs
# LOCAL_RANK: 0 - CUDA_VISIBLE_DEVICES: [0]
# Epoch 500/500: 100%|██████████| 500/500 [02:51<00:00, 2.91it/s, loss=-9.3e+03, v_num=1]
# `Trainer.fit` stopped: `max_epochs=500` reached.
# Epoch 500/500: 100%|██████████| 500/500 [02:51<00:00, 2.91it/s, loss=-9.3e+03, v_num=1]
import torch
def add_velovi_outputs_to_adata(adata, vae):
latent_time = vae.get_latent_time(n_samples=25)
velocities = vae.get_velocity(n_samples=25, velo_statistic="mean")
t = latent_time
scaling = 20 / t.max(0)
adata.layers["velocity"] = velocities / scaling
adata.layers["latent_time_velovi"] = latent_time
adata.var["fit_alpha"] = vae.get_rates()["alpha"] / scaling
adata.var["fit_beta"] = vae.get_rates()["beta"] / scaling
adata.var["fit_gamma"] = vae.get_rates()["gamma"] / scaling
adata.var["fit_t_"] = (
torch.nn.functional.softplus(vae.module.switch_time_unconstr)
.detach()
.cpu()
.numpy()
) * scaling
adata.layers["fit_t"] = latent_time.values * scaling[np.newaxis, :]
adata.var['fit_scaling'] = 1.0
add_velovi_outputs_to_adata(adata, vae)
scv.tl.velocity_graph(adata, n_jobs=8)
scv.pl.velocity_embedding_stream(adata, basis="umap")
# computing velocity graph (using 8/12 cores)
# 0%| | 0/2531 [00:00<?, ?cells/s]
# finished (0:00:08) --> added
# 'velocity_graph', sparse matrix with cosine correlations (adata.uns)
# computing velocity embedding
# finished (0:00:00) --> added
# 'velocity_umap', embedded velocity vectors (adata.obsm)

Интересно, что модель глубокого обучения уловила рециркуляцию альфа- и эпсилон-клеток.
scv.tl.latent_time(adata)
adata.obs['latent_time_velovi']=adata.obs['latent_time'].copy()
# computing latent time using root_cells as prior
# finished (0:00:00) --> added
# 'latent_time', shared time (adata.obs)
adata.obs['velocity_pseudotime_velovi']=adata.obs['velocity_pseudotime'].copy()
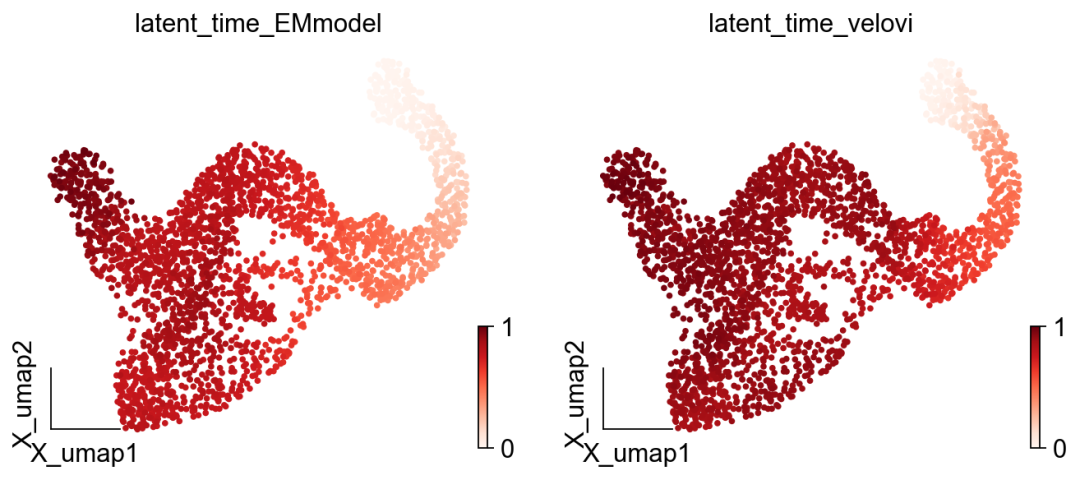
Чтобы сравнить различные значения псевдовремени модели, мы можем использовать внедрение для визуализации задержки.
ov.pl.embedding(adata,basis='X_umap',
color=['latent_time_EMmodel','latent_time_velovi'],
frameon='small',cmap='Reds')
9. Меры предосторожности
Чтобы понять, применим ли анализ скорости РНК к данному набору данных, отметим следующие моменты:
- Чтобы сделать вывод о скорости РНК, временной масштаб изучаемого процесса развития должен быть сопоставим с периодом полураспада молекулы РНК. Например, это требование соблюдается при развитии эндокринной системы поджелудочной железы, но не при хронических заболеваниях, таких как болезнь Альцгеймера или Паркинсона. Аналогично, анализ скорости РНК не подходит для стационарных систем, в которых отсутствует какой-либо обмен (например, мононуклеарные клетки периферической крови).
- Только если предположение базовой Модели выполняется (приблизительно),Только тогда можно будет точно и надежно определить скорость РНК. Чтобы проверить эти предположения,Фазовые диаграммы можно изучить, чтобы убедиться, что они принимают ожидаемую миндалевидную форму. Если ген содержит несколько различных динамик,Анализ скорости РНК следует применять с осторожностью.,и, возможно, подгруппировать данные в одну линию.
- Традиционно,Векторы скорости РНК высокой размерности визуализируются путем проецирования их на низкоразмерное представление данных. Такой подход к проверке предположений может быть неверным и вводить в заблуждение.,Потому что прогнозируемая скорость потока сильно зависит от (1) количества включенных генов и (2) выбранных параметров построения. также,Качество проекции ухудшается на границах низкоразмерных вложений.



Неразрушающее увеличение изображений одним щелчком мыши, чтобы сделать их более четкими артефактами искусственного интеллекта, включая руководства по установке и использованию.
Копикодер: этот инструмент отлично работает с Cursor, Bolt и V0! Предоставьте более качественные подсказки для разработки интерфейса (создание навигационного веб-сайта с использованием искусственного интеллекта).
Новый бесплатный RooCline превосходит Cline v3.1? ! Быстрее, умнее и лучше вилка Cline! (Независимое программирование AI, порог 0)

Разработав более 10 проектов с помощью Cursor, я собрал 10 примеров и 60 подсказок.
Я потратил 72 часа на изучение курсорных агентов, и вот неоспоримые факты, которыми я должен поделиться!

Идеальная интеграция Cursor и DeepSeek API
DeepSeek V3 снижает затраты на обучение больших моделей
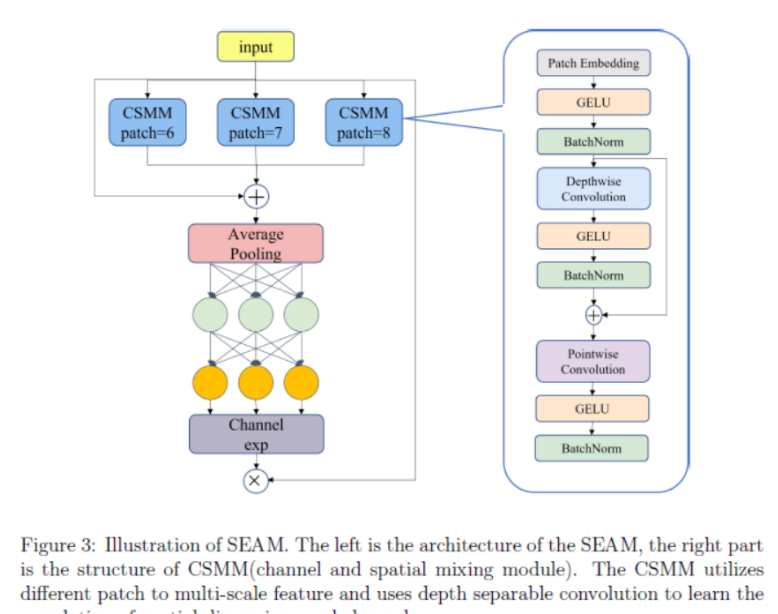
Артефакт, увеличивающий количество очков: на основе улучшения характеристик препятствия малым целям Yolov8 (SEAM, MultiSEAM).

DeepSeek V3 раскручивался уже три дня. Сегодня я попробовал самопровозглашенную модель «ChatGPT».

Open Devin — инженер-программист искусственного интеллекта с открытым исходным кодом, который меньше программирует и больше создает.
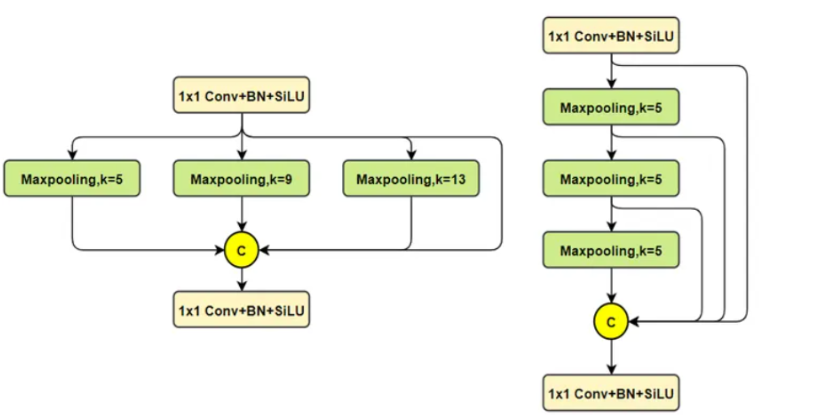
Эксклюзивное оригинальное улучшение YOLOv8: собственная разработка SPPF | SPPF сочетается с воспринимаемой большой сверткой ядра UniRepLK, а свертка с большим ядром + без расширения улучшает восприимчивое поле

Популярное и подробное объяснение DeepSeek-V3: от его появления до преимуществ и сравнения с GPT-4o.

9 основных словесных инструкций по доработке академических работ с помощью ChatGPT, эффективных и практичных, которые стоит собрать
Вызовите deepseek в vscode для реализации программирования с помощью искусственного интеллекта.
Познакомьтесь с принципами сверточных нейронных сетей (CNN) в одной статье (суперподробно)
50,3 тыс. звезд! Immich: автономное решение для резервного копирования фотографий и видео, которое экономит деньги и избавляет от беспокойства.
Cloud Native|Практика: установка Dashbaord для K8s, графика неплохая
Краткий обзор статьи — использование синтетических данных при обучении больших моделей и оптимизации производительности
MiniPerplx: новая поисковая система искусственного интеллекта с открытым исходным кодом, спонсируемая xAI и Vercel.
Конструкция сервиса Synology Drive сочетает проникновение в интрасеть и синхронизацию папок заметок Obsidian в облаке.
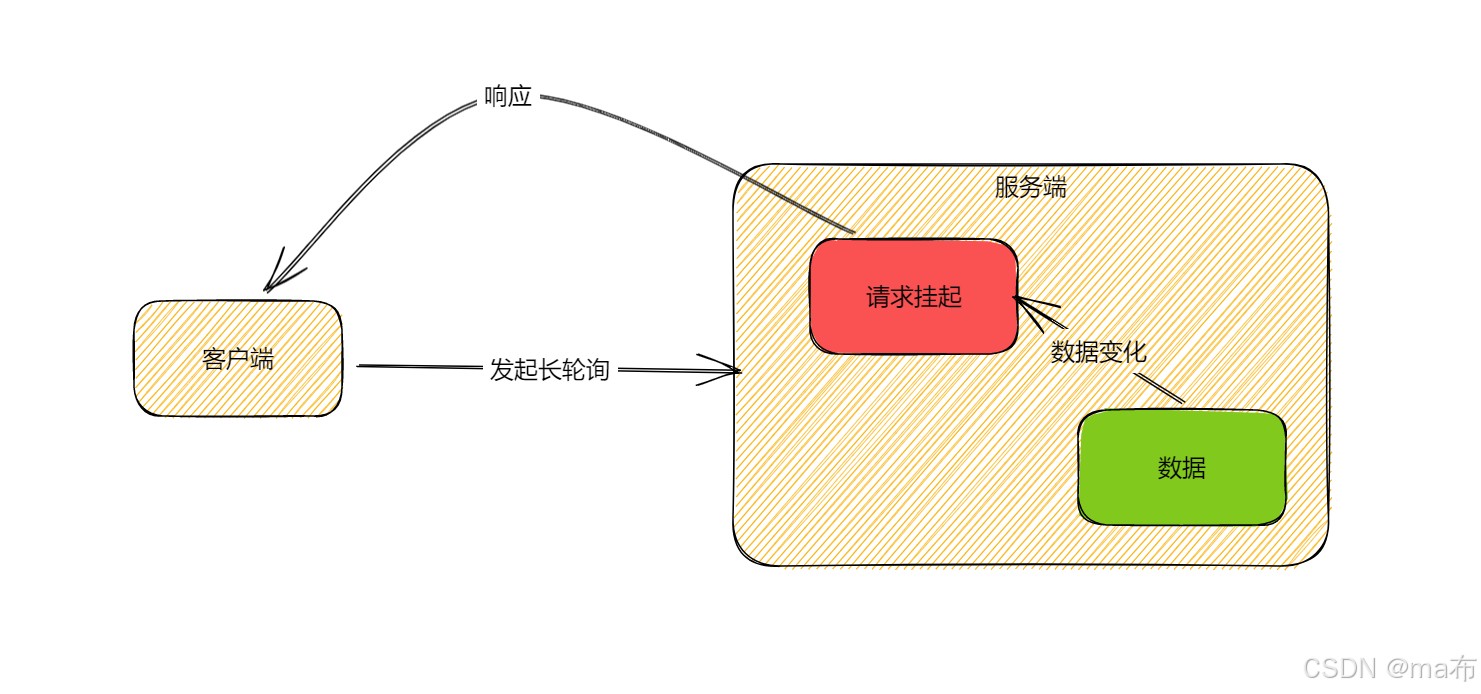
Центр конфигурации————Накос
Начинаем с нуля при разработке в облаке Copilot: начать разработку с минимальным использованием кода стало проще

[Серия Docker] Docker создает мультиплатформенные образы: практика архитектуры Arm64

Обновление новых возможностей coze | Я использовал coze для создания апплета помощника по исправлению домашних заданий по математике

Советы по развертыванию Nginx: практическое создание статических веб-сайтов на облачных серверах
Feiniu fnos использует Docker для развертывания личного блокнота Notepad

Сверточная нейронная сеть VGG реализует классификацию изображений Cifar10 — практический опыт Pytorch
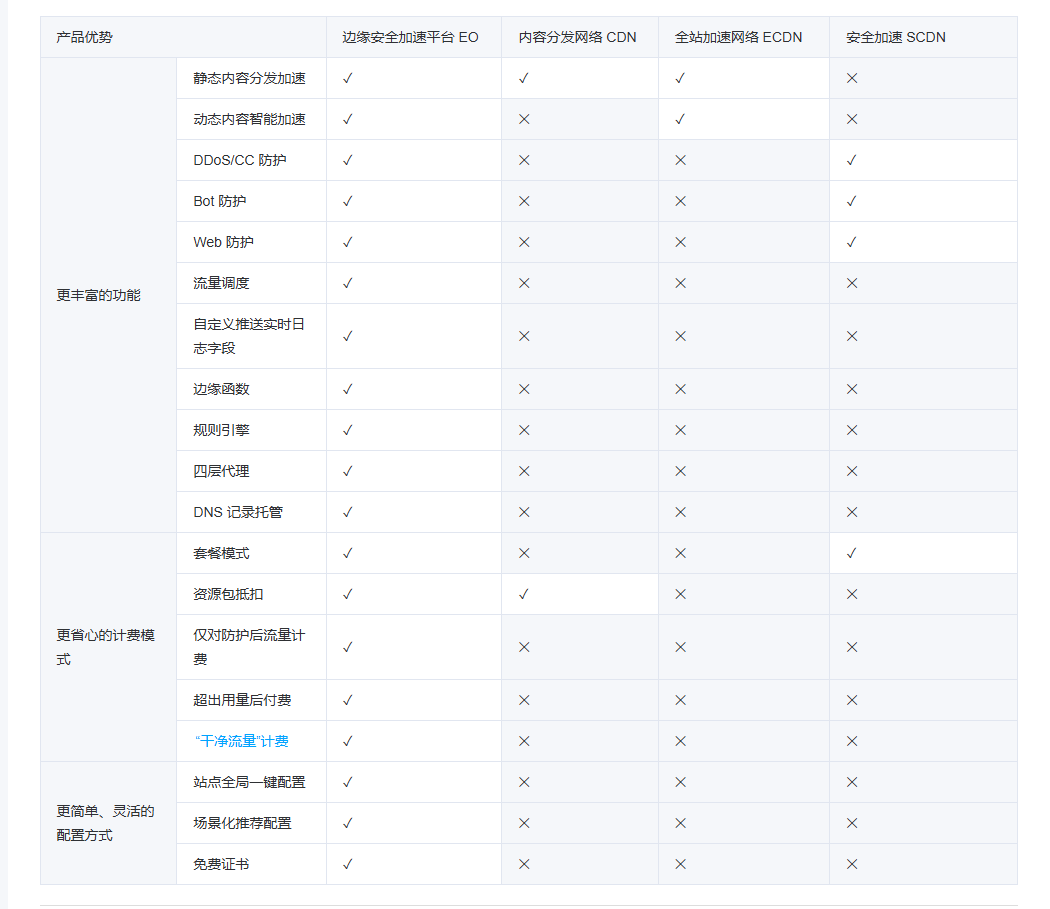
Начало работы с EdgeonePages — новым недорогим решением для хостинга веб-сайтов
[Зона легкого облачного игрового сервера] Управление игровыми архивами