Легко обрабатывать этот набор данных из 1,3 миллиона отдельных ячеек в R
Эта заметка будет включена в альбом «Single Cell 2024» общедоступной учетной записи «Дерево навыков Шэнсинь», а все наши учебные пособия, начиная с 2024 года, основаны на версии Seurat V5. Ранее мы демонстрировали, как читать отдельные ячейки в разных форматах. форматы файла данных транскриптома, как показано ниже:
- Первая попытка версии Seurat V5
- Используйте Seurat v5 для чтения нескольких 10-кратных матриц транскриптома одной клетки
- Использование версии Seurat v5 для чтения нескольких одноячеечных проектов, которые не являются стандартными файлами 10x.
Потому что эта версия Seurat V5 по-прежнему имеет некоторые преимущества. Например, она может легко обрабатывать набор данных из 1,3 миллиона ячеек. Вам необходимо обратиться к трем данным на официальном сайте Seurat:
- https://bnprks.github.io/BPCells/articles/pbmc3k.html
- https://satijalab.org/seurat/articles/seurat5_sketch_analysis
- https://satijalab.org/seurat/articles/seurat5_bpcells_interaction_vignette
Пакет R BPCells может считывать и сохранять набор данных из 1,3 миллиона отдельных ячеек в дисковом хранилище, а метод Sketching может выполнять выборку из набора данных из 1,3 миллиона отдельных ячеек, сохраняя при этом характеристики набора данных.
Установите пакет BPCells
Первым шагом является установка и загрузка необходимых пакетов:
remotes::install_github("bnprks/BPCells")
# https://api.github.com/repos/bnprks/BPCells/tarball/HEAD
library(BPCells)
library(Seurat)
library(SeuratObject)
library(SeuratDisk)
library(Azimuth)
# needs to be set for large dataset analysis
options(future.globals.maxSize = 1e9)
Поскольку пакет BPCells до сих пор хранится на github, камнем преткновения в первую очередь является сеть. Тогда, если вы не используете операционную систему Windows, вам, скорее всего, не хватает hdf5;
Searching for hdf5 in a conda env...
Unable to locate libhdf5. Please install manually or edit compiler flags.
ERROR: configuration failed for package ‘BPCells’
В разных операционных системах есть соответствующие решения, которые заключаются в установке hdf5:
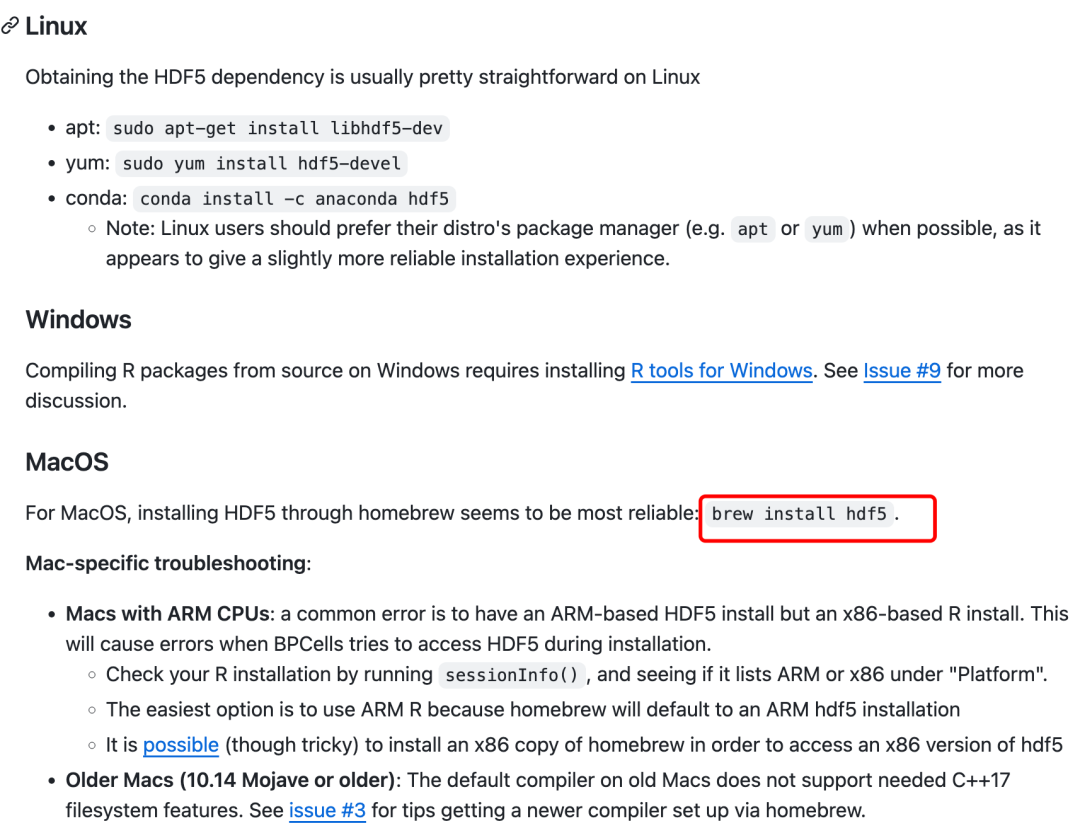
Соответствующее решение
После того, как все будет сделано, вы можете легко загрузить и использовать пакет BPCells R.
Просмотр и чтение набора данных из 1,3 миллиона отдельных ячеек (файл h5)
Набор данных по 1,3 миллионам отдельных клеток предоставлен компанией 10x на ее официальном сайте. Ссылка: https://support.10xgenomics.com/single-cell-gene-expression/datasets/1.3.0/1M_neurons.
Вам нужно просто войти в систему, чтобы увидеть ссылку для скачивания. Вы можете видеть, что размер файла матрицы выражений близок к 4G: https://cf.10xgenomics.com/samples/cell-exp/1.3.0/1M_neurons/. 1M_neurons_filtered_gene_bc_matrices_h5.h5
Ссылка для скачивания
Далее давайте посмотрим, как пакет BPCells R работает с этим файлом h5.
brain.data <- open_matrix_10x_hdf5(
path = "1M_neurons_filtered_gene_bc_matrices_h5.h5"
)
# Write the matrix to a directory
write_matrix_dir(
mat = brain.data,
dir = 'brain_counts')
# Now that we have the matrix on disk, we can load it
brain.mat <- open_matrix_dir(dir = "brain_counts")
brain.mat <- Azimuth:::ConvertEnsembleToSymbol(mat = brain.mat, species = "mouse")
# Create Seurat Object
brain <- CreateSeuratObject(counts = brain.mat)
Видно, что считывание матрицы экспрессии одноклеточного транскриптома из файла h5 в R, чтобы превратить ее в разреженную матрицу, состоит в основном из трех шагов. После простого преобразования идентификатора гена в Seurat можно создать объект Seurat. Ниже приводится объяснение каждого шага:
open_matrix_10x_hdf5: из 10x Genomics из HDF5 Считайте одноклеточный транскриптом из данных файла. Этот документ обычно содержит информацию о секвенировании отдельных клеток и необработанную информацию о подсчете.write_matrix_dir: Запишите прочитанные данные одноклеточного транскриптома в указанный каталог. Этот шаг может заключаться в сохранении файла на диске для последующего анализа.open_matrix_dir: Считайте данные одноклеточного транскриптома из указанного каталога. Этот шаг необходим для того, чтобы файл был записан на диск и его можно было легко использовать для последующих операций.Azimuth:::ConvertEnsembleToSymbol: использовать Azimuth В пакете функций ген из Ensembl Логотипы преобразуются в символы. Ансамбль Это метод идентификации гена, который может быть легче понять и использовать, преобразованный в символы.CreateSeuratObject: использовать Seurat изфункция в пакете создает ее на основе заданных данных транскриптома Seurat объект。Seurat это популярный метод анализа транскриптома отдельных клеток. R Сумка.
Целью всего процесса является чтение, хранение, преобразование необработанных данных транскриптома одной клетки и, наконец, создание объекта Сёра для последующего анализа одной клетки. Например, вы можете просто посмотреть на уровни экспрессии некоторых генов:
VlnPlot(brain, features = c("Sox10", "Slc17a7", "Aif1"), ncol = 3,
layer = "counts", alpha = 0.1)
# We then normalize and visualize again
brain <- NormalizeData(brain, normalization.method = "LogNormalize")
VlnPlot(brain, features = c("Sox10", "Slc17a7", "Aif1"), ncol = 3,
layer = "data", alpha = 0.1)
Исходный объект Сёра имеет только матрицу счетчиков (чистые целые числа), но после обработки функцией NormalizeData появляется информация о данных. Вы можете увидеть разницу, визуализировав ее до и после:
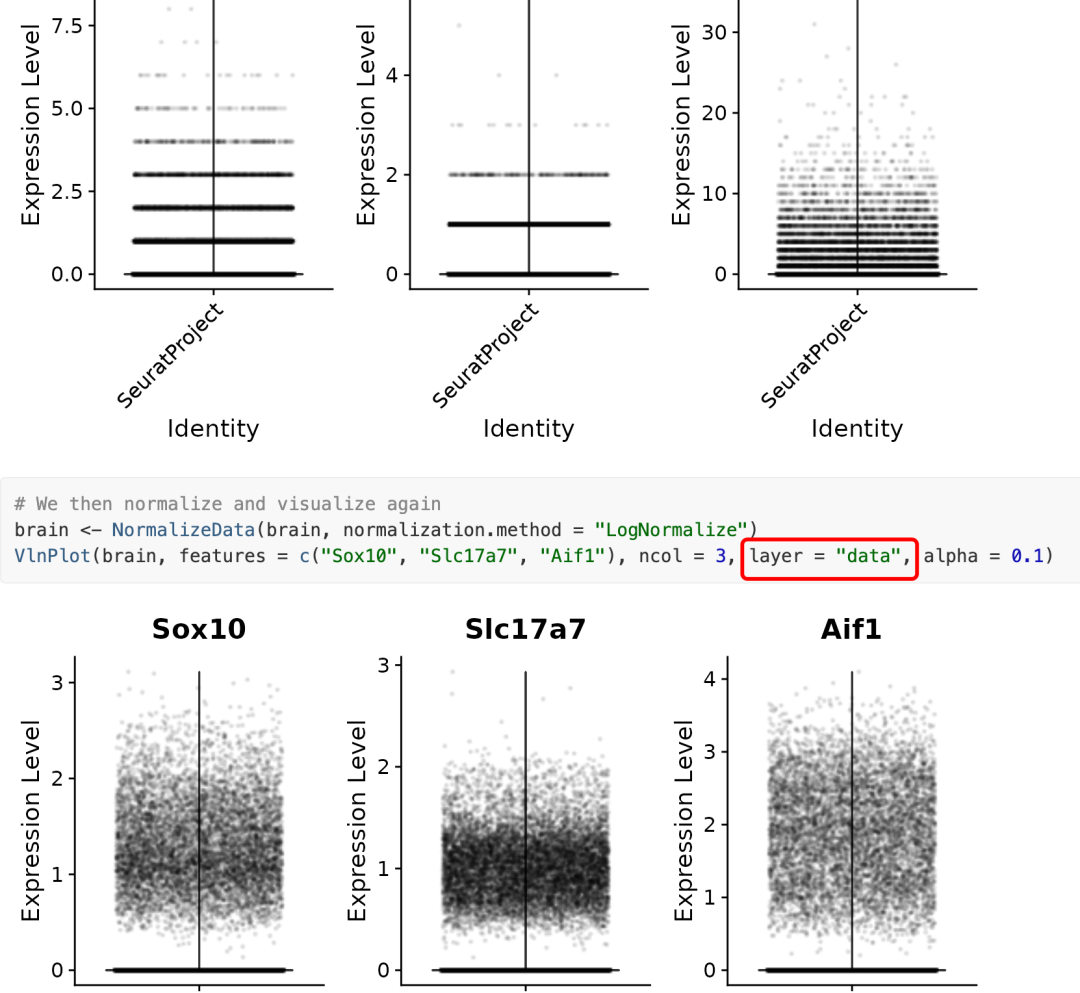
После обработки функцией NormalizeData
Этот объект Seurat также можно сохранить обычным способом в виде файла rds на языке R, как показано ниже:
SaveSeuratRds(
object = brain,
file = "1p3_million_mouse_brain.rds"
)
Позже вы можете напрямую использовать readRDS для чтения сохраненного файла rds на языке R, не начиная с файла матрицы выражений одной ячейки в формате h5.
Выборка и последующая кластеризация с уменьшением размерности
Хотя ранее мы использовали пакет BPCells R для чтения матрицы экспрессии одноклеточного транскриптома в файле h5 в R и создали объект Seurat, непосредственное выполнение последующей кластеризации с уменьшением размерности на этом объекте Seurat по-прежнему требует каждого. Ресурсы, потребляемые на каждом этапе, ужасны. . На данный момент вам необходимо использовать метод эскиза для выборки из 1,3 миллиона наборов данных с одной ячейкой, но при этом сохранить характеристики набора данных. Сначала прочитайте файл rds на языке R, сохраненный ранее:
# Read the Seurat object, which contains 1.3M cells stored on-disk as part of the 'RNA' assay
obj <- readRDS("1p3_million_mouse_brain.rds")
obj
## An object of class Seurat
## 27282 features across 1306127 samples within 1 assay
## Active assay: RNA (27282 features, 0 variable features)
## 1 layer present: counts
# Note that since the data is stored on-disk, the object size easily fits in-memory (<1GB)
format(object.size(obj), units = "Mb")
## [1] "596.2 Mb"
Затем образец:
obj <- NormalizeData(obj)
obj <- FindVariableFeatures(obj)
obj <- SketchData(
object = obj,
ncells = 50000,
method = "LeverageScore",
sketched.assay = "sketch"
)
obj
Далее просто выполните уменьшение размерности и кластеризацию;
DefaultAssay(obj) <- "sketch"
obj <- FindVariableFeatures(obj)
obj <- ScaleData(obj)
obj <- RunPCA(obj)
obj <- FindNeighbors(obj, dims = 1:50)
obj <- FindClusters(obj, resolution = 2)
obj <- RunUMAP(obj, dims = 1:50, return.model = T)
DimPlot(obj, label = T, label.size = 3, reduction = "umap") + NoLegend()
Видно, что хотя после выборки это подмножество, количество ячеек на самом деле очень значительное:
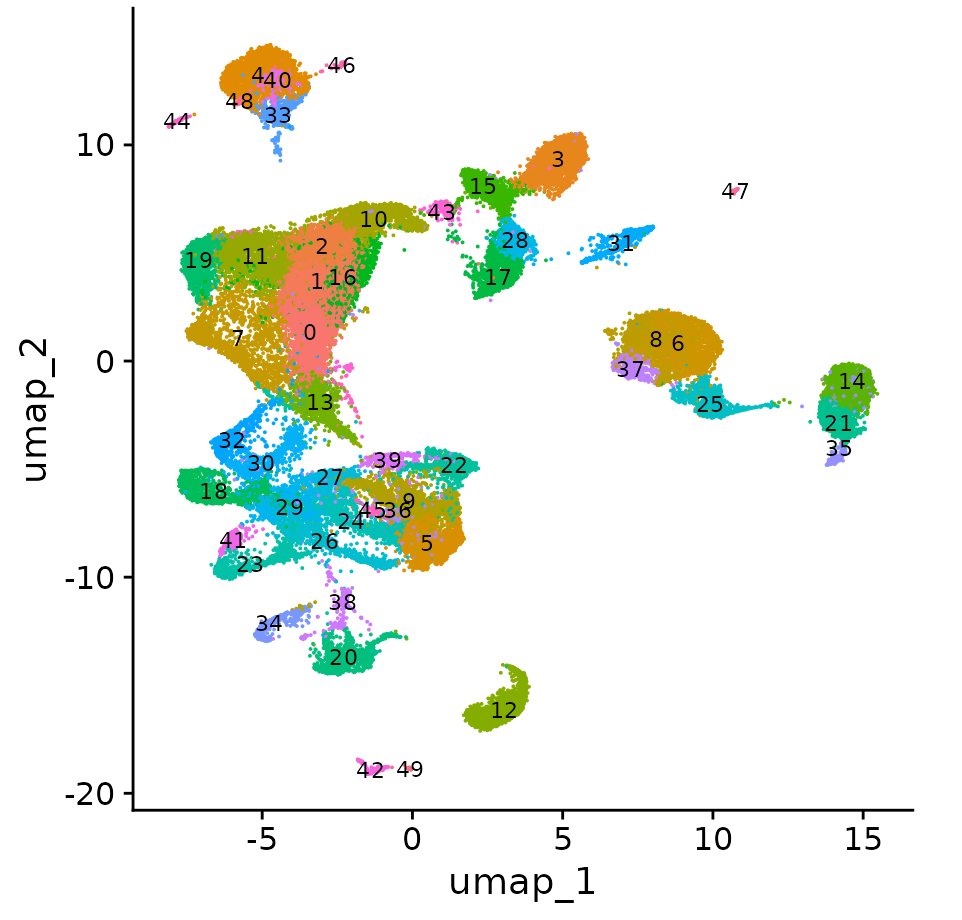
Количество клеток также значительное.
Поскольку это мозг мыши, мы можем кратко рассмотреть общие гены с высокой экспрессией, специфичные для каждой отдельной субпопуляции клеток, основываясь на биологических знаниях:
FeaturePlot(
object = obj,
features = c(
"Igfbp7", "Neurod6", "Dlx2", "Gad2",
"Eomes", "Vim", "Reln", "Olig1", "C1qa"
),
ncol = 3
)
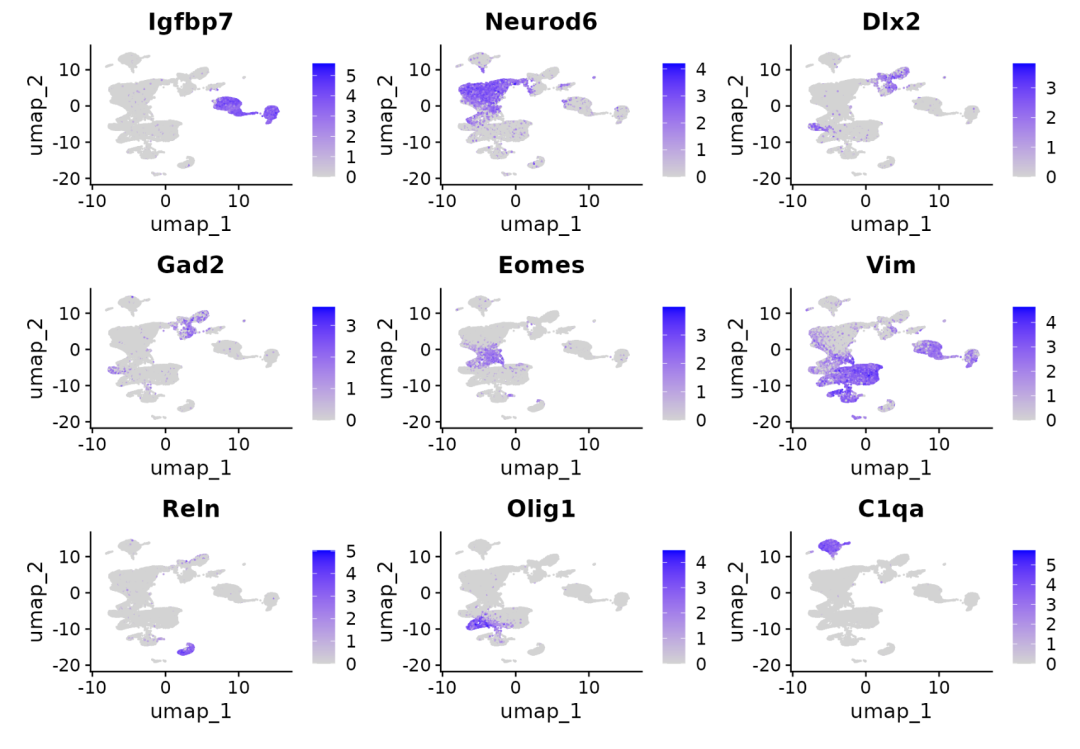
image-20240104144750156
Результаты кластеризации с уменьшением размерности подмножества можно сопоставить с предыдущим полным набором данных, который составляет 1,3 миллиона отдельных ячеек. На этом этапе используйте функцию ProjectData:
obj <- ProjectData(
object = obj,
assay = "RNA",
full.reduction = "pca.full",
sketched.assay = "sketch",
sketched.reduction = "pca",
umap.model = "umap",
dims = 1:50,
refdata = list(cluster_full = "seurat_clusters")
)
# now that we have projected the full dataset, switch back to analyzing all cells
DefaultAssay(obj) <- "RNA"
DimPlot(obj, label = T, label.size = 3, reduction = "full.umap", group.by = "cluster_full", alpha = 0.1) + NoLegend()
Как видите, по сути это тот же UMAP-граф, разница в том, что количество ячеек гораздо больше:
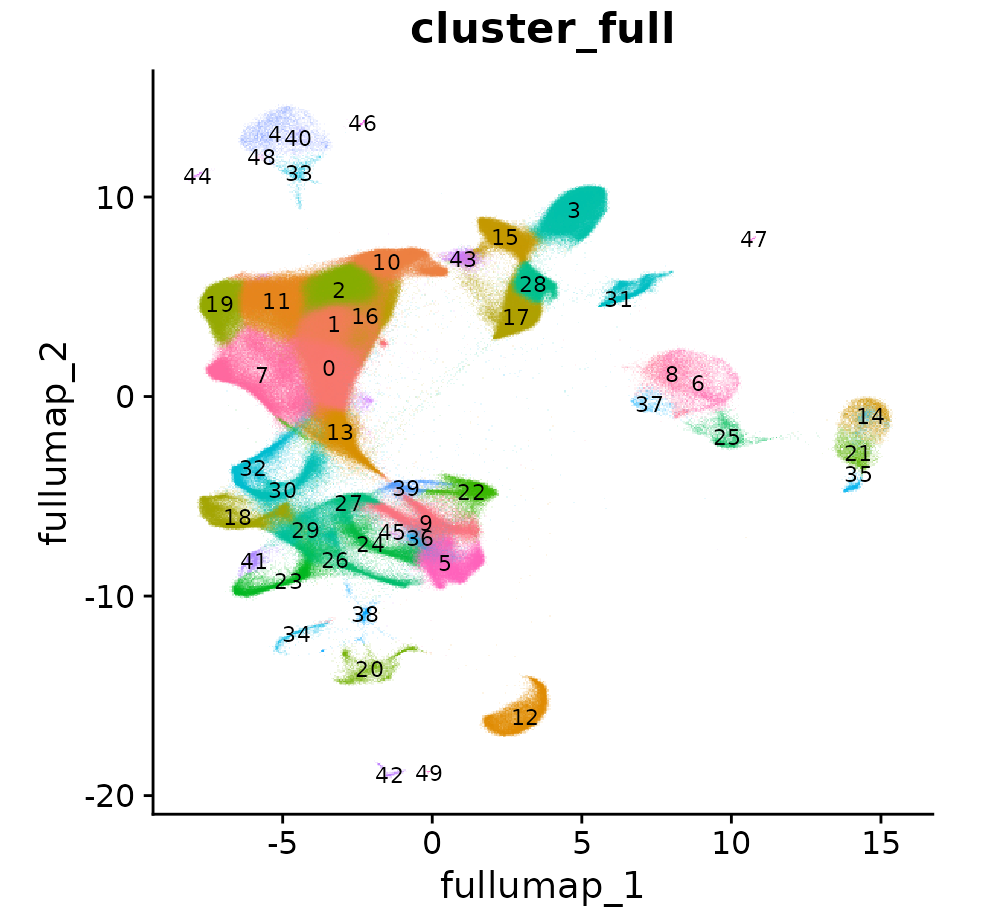
Похожая графика umap
Стоит отметить, что на данный момент в нашем объекте Сёра есть два разных анализа, которые необходимо тщательно различать:
# visualize gene expression on the sketched cells (fast) and the full dataset (slower)
# switch to analyzing the sketched dataset (in-memory)
DefaultAssay(obj) <- "sketch"
x1 <- FeaturePlot(obj, "C1qa")
# switch to analyzing the full dataset (on-disk)
DefaultAssay(obj) <- "RNA"
x2 <- FeaturePlot(obj, "C1qa")
x1 | x2
Пять распространенных визуализаций Сёра, о которых мы упоминали ранее, можно проверить с помощью этих двух разных тестов.
После взятия подмножества нет необходимости проводить выборку
Предыдущий набор данных составляет 1,3 миллиона отдельных ячеек, но после кластеризации с уменьшением размерности, если нас интересуют только некоторые подгруппы, например, выбор 2, 15, 18, 28, 40 подгрупп из UMAP выше, фактически количество ячеек составляет всего около 200 000. Вы можете напрямую сделать его обычным объектом Сёра, который хранит в памяти разреженную матрицу без выборки, а затем выполнять обычную кластеризацию с уменьшением размерности;
# subset cells in these clusters. Note that the data remains on-disk after subsetting
obj.sub <- subset(obj, subset = cluster_full %in% c(2, 15, 18, 28, 40))
DefaultAssay(obj.sub) <- "RNA"
# now convert the RNA assay (previously on-disk) into an in-memory representation (sparse Matrix)
# we only convert the data layer, and keep the counts on-disk
obj.sub[["RNA"]]$data <- as(obj.sub[["RNA"]]$data, Class = "dgCMatrix")
# recluster the cells
obj.sub <- FindVariableFeatures(obj.sub)
obj.sub <- ScaleData(obj.sub)
obj.sub <- RunPCA(obj.sub)
obj.sub <- RunUMAP(obj.sub, dims = 1:30)
obj.sub <- FindNeighbors(obj.sub, dims = 1:30)
obj.sub <- FindClusters(obj.sub)
DimPlot(obj.sub, label = T, label.size = 3) + NoLegend()
Наши обычные компьютеры или серверы могут содержать около 200 000 ячеек. Вы можете попробовать посмотреть на конкретные гены различных субпопуляций отдельных клеток в областях мозга, которые мы собрали ранее, чтобы увидеть, эффективны ли они в этом наборе данных. Посмотреть подробности :Сравнение клеточных субпопуляций и их маркерных генов из двух групп одноклеточных ядерных транскриптомов двух нейродегенеративных заболеваний,Я перечислил некоторые гены:
astrocytes = c("AQP4", "ADGRV1", "GPC5", "RYR3")
endothelial = c("CLDN5", "ABCB1", "EBF1")
excitatory = c("CAMK2A", "CBLN2", "LDB2")
inhibitory = c("GAD1", "LHFPL3", "PCDH15")
microglia = c("C3", "LRMDA", "DOCK8")
oligodendrocytes = c("MBP", "PLP1", "ST18")
OPC='Tnr,Igsf21,Neu4,Gpr17'
Ependymal='Cfap126,Fam183b,Tmem212,pifo,Tekt1,Dnah12'
pericyte=c( 'DCN', 'LUM', 'GSN' ,'FGF7','MME', 'ACTA2','RGS5')
# Ниже представлены 4 типа нервных клеток.
# excitatory (SLC17A6),
# inhibitory (GAD2),
# GABAergic (GAD2/GRIK1),
# dopaminergic neurons(TH)
Среди них нейроны также можно подразделить на:
- motoneurons (MN),
- excitatory dorsal neurons (ExDorsal),
- inhibitory dorsal neurons (InhDorsal),
- excitatory mid and ventral neurons (ExMV),
- inhibitory or mixed mid and ventral neurons (InhMV).
Он также может быть более подробным, в зависимости от соответствующего биологического фона.



Неразрушающее увеличение изображений одним щелчком мыши, чтобы сделать их более четкими артефактами искусственного интеллекта, включая руководства по установке и использованию.
Копикодер: этот инструмент отлично работает с Cursor, Bolt и V0! Предоставьте более качественные подсказки для разработки интерфейса (создание навигационного веб-сайта с использованием искусственного интеллекта).
Новый бесплатный RooCline превосходит Cline v3.1? ! Быстрее, умнее и лучше вилка Cline! (Независимое программирование AI, порог 0)

Разработав более 10 проектов с помощью Cursor, я собрал 10 примеров и 60 подсказок.
Я потратил 72 часа на изучение курсорных агентов, и вот неоспоримые факты, которыми я должен поделиться!

Идеальная интеграция Cursor и DeepSeek API
DeepSeek V3 снижает затраты на обучение больших моделей
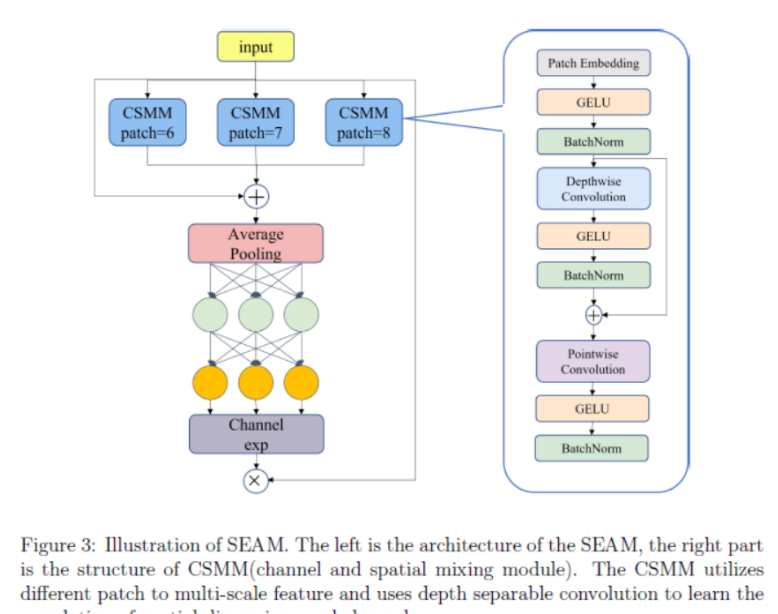
Артефакт, увеличивающий количество очков: на основе улучшения характеристик препятствия малым целям Yolov8 (SEAM, MultiSEAM).

DeepSeek V3 раскручивался уже три дня. Сегодня я попробовал самопровозглашенную модель «ChatGPT».

Open Devin — инженер-программист искусственного интеллекта с открытым исходным кодом, который меньше программирует и больше создает.
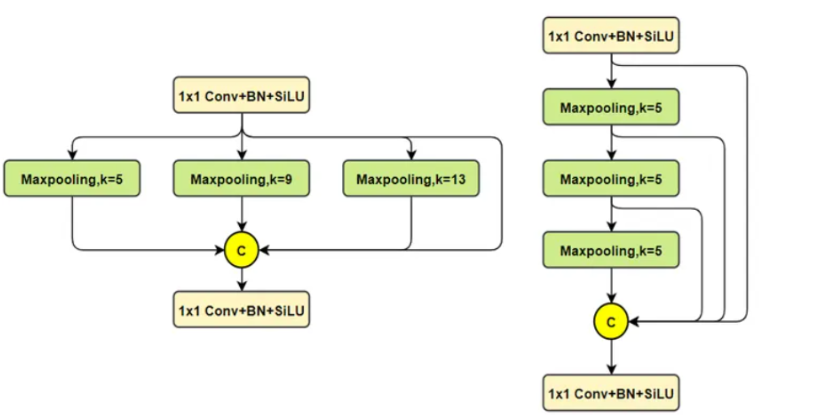
Эксклюзивное оригинальное улучшение YOLOv8: собственная разработка SPPF | SPPF сочетается с воспринимаемой большой сверткой ядра UniRepLK, а свертка с большим ядром + без расширения улучшает восприимчивое поле

Популярное и подробное объяснение DeepSeek-V3: от его появления до преимуществ и сравнения с GPT-4o.

9 основных словесных инструкций по доработке академических работ с помощью ChatGPT, эффективных и практичных, которые стоит собрать
Вызовите deepseek в vscode для реализации программирования с помощью искусственного интеллекта.
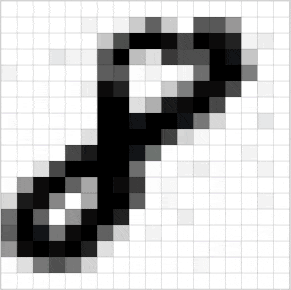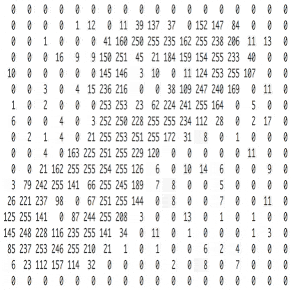
Познакомьтесь с принципами сверточных нейронных сетей (CNN) в одной статье (суперподробно)
50,3 тыс. звезд! Immich: автономное решение для резервного копирования фотографий и видео, которое экономит деньги и избавляет от беспокойства.
Cloud Native|Практика: установка Dashbaord для K8s, графика неплохая
Краткий обзор статьи — использование синтетических данных при обучении больших моделей и оптимизации производительности
MiniPerplx: новая поисковая система искусственного интеллекта с открытым исходным кодом, спонсируемая xAI и Vercel.
Конструкция сервиса Synology Drive сочетает проникновение в интрасеть и синхронизацию папок заметок Obsidian в облаке.
Центр конфигурации————Накос
Начинаем с нуля при разработке в облаке Copilot: начать разработку с минимальным использованием кода стало проще

[Серия Docker] Docker создает мультиплатформенные образы: практика архитектуры Arm64

Обновление новых возможностей coze | Я использовал coze для создания апплета помощника по исправлению домашних заданий по математике

Советы по развертыванию Nginx: практическое создание статических веб-сайтов на облачных серверах
Feiniu fnos использует Docker для развертывания личного блокнота Notepad

Сверточная нейронная сеть VGG реализует классификацию изображений Cifar10 — практический опыт Pytorch
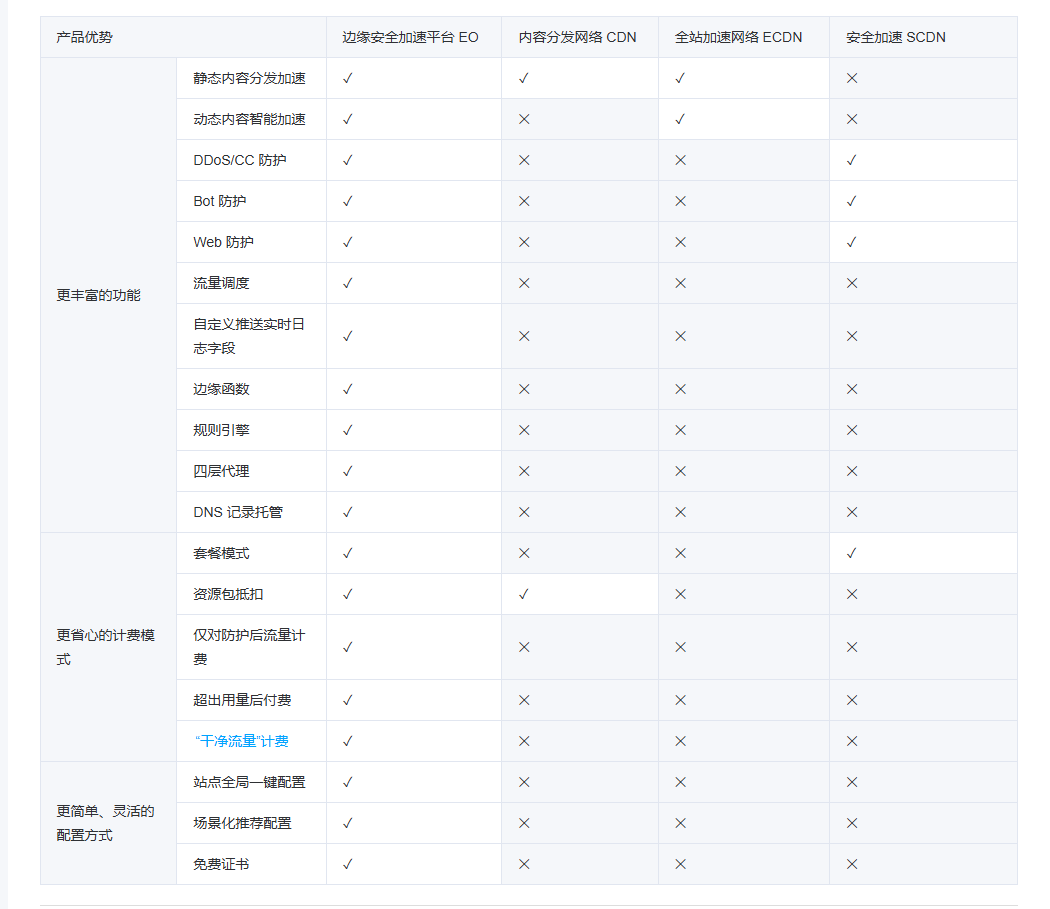
Начало работы с EdgeonePages — новым недорогим решением для хостинга веб-сайтов
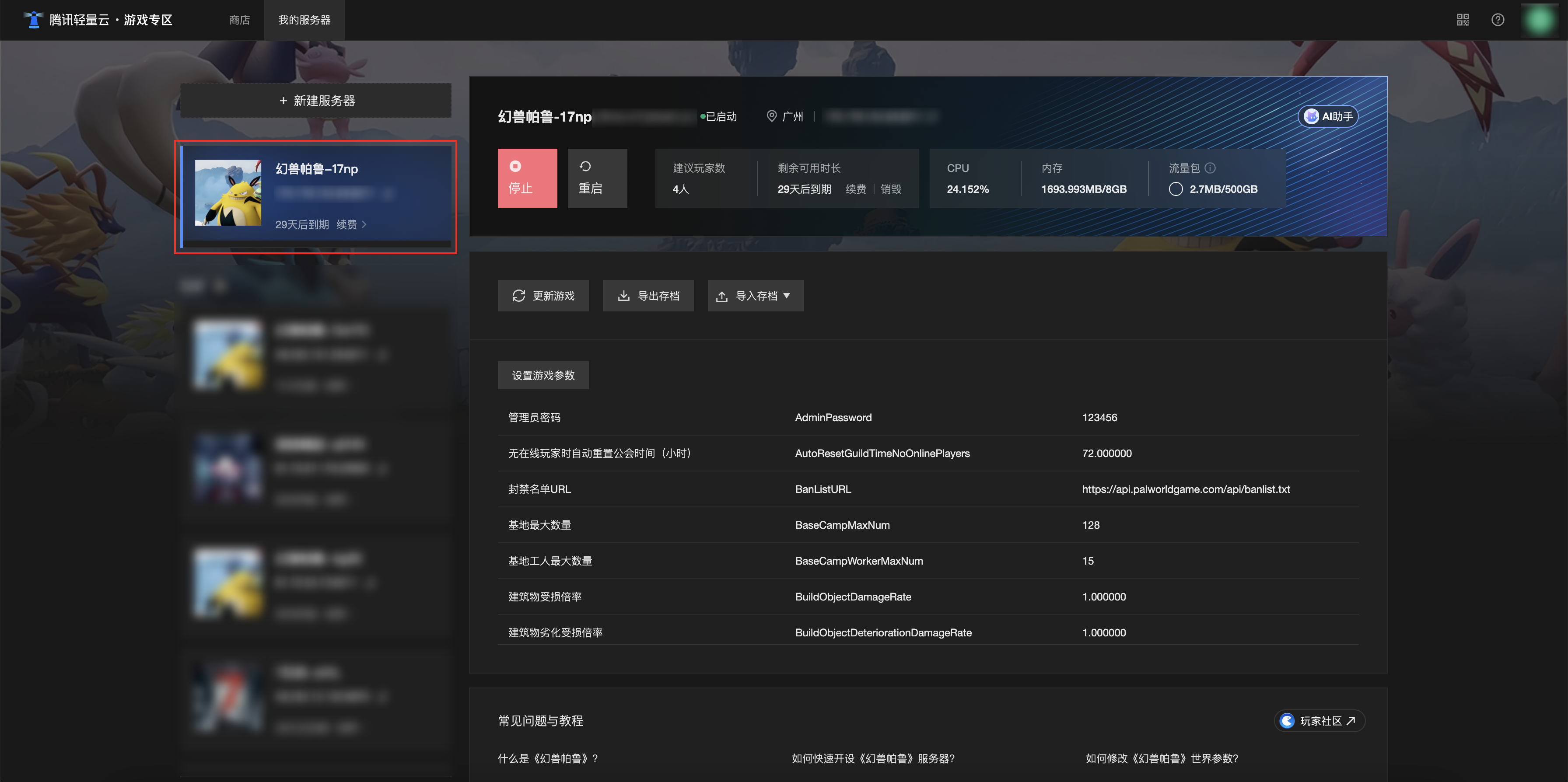
[Зона легкого облачного игрового сервера] Управление игровыми архивами