Коллекция Banyan Tree — введение, установка и использование RoseTTAFold All-Atom

Предисловие
Я следил за последней статьей AF, которая официально называется AlphaFold-последняя. Она может предсказывать сложную структуру белков и малых молекул, белков и нуклеиновых кислот. Это атака с уменьшением размерности на традиционные методы сложного прогнозирования. Единственный недостаток: по слухам, Deepmind не планирует открывать исходный код. Ну да ладно, это мой недостаток. Подробности о статье ниже.
Banyan Set – обновления AlphaFold
Ну и Розетта тоже дала полноатомное решение. Статья опубликована 7, 24 марта, и до сих пор горячо. RoseTTAFold All-Atom (RFAA) может предсказывать белки, нуклеиновые кислоты, малые молекулы, металлы и ковалентно модифицированные сборки и делает его открытым исходным кодом.
Сейчас мне больше любопытно, живем ли мы в эпоху, когда ИИ может предоставить окончательное решение, или мы живем в эпоху, когда все работают вместе, чтобы строить и улучшать.
Введение
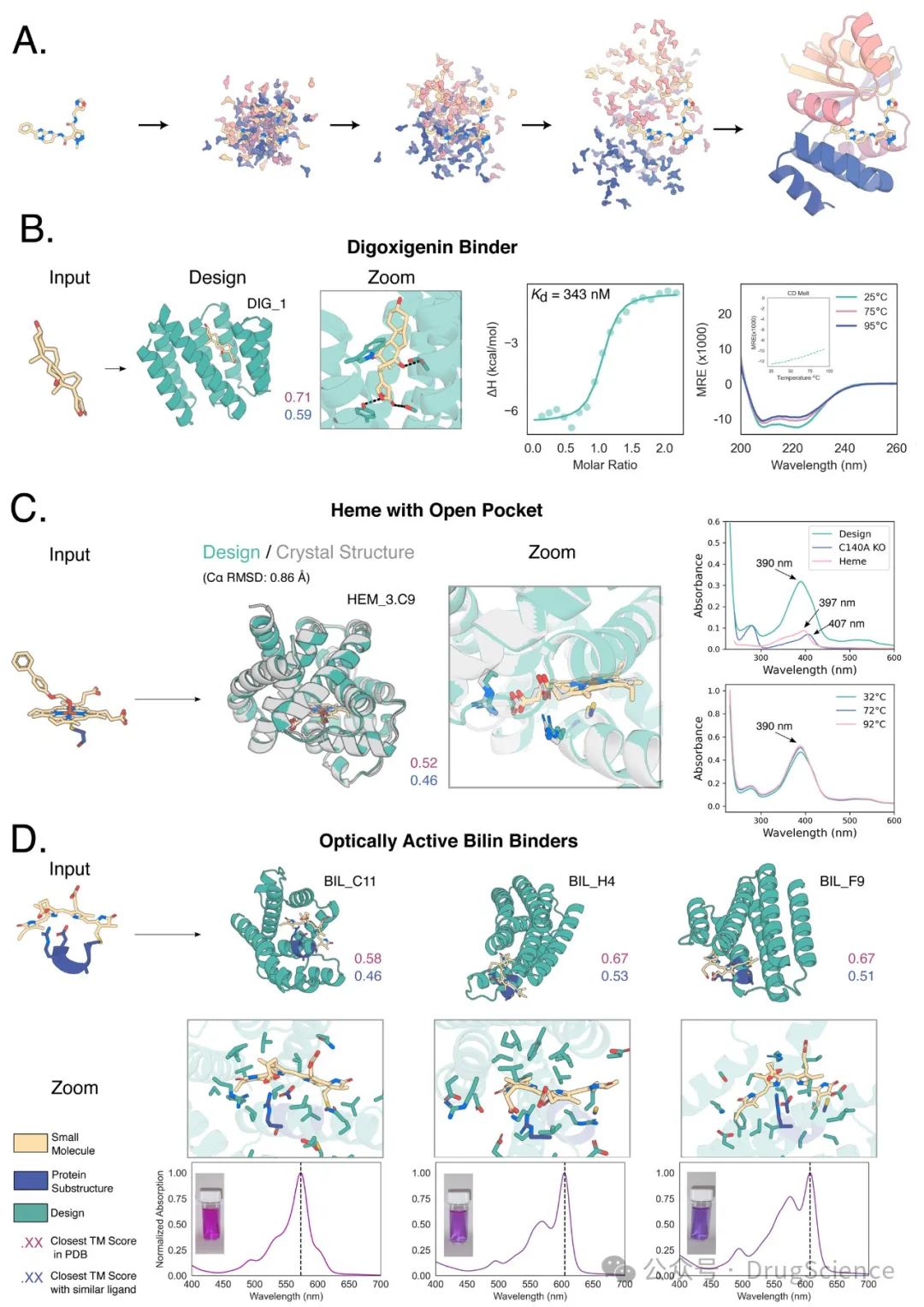
Методы глубокого обучения произвели революцию в области прогнозирования и проектирования структуры белков. В этой статье представлены две модели: 1. RoseTTAFold All-Atom (RFAA), которая может моделировать сборки, содержащие белки, малые молекулы, нуклеиновые кислоты, металлы и ковалентные модификации. 2. RFdiffusionAA, которая выполняет задачи снижения шума. Был получен RFdiffusionAA, который может строить белковые структуры вокруг небольших молекул.
Авторы также провели экспериментальную проверку и разработали белки, которые могут связывать дигоксигенин, белки, которые могут связывать гем, и белки, которые могут связывать светособирающую молекулу биливердин.
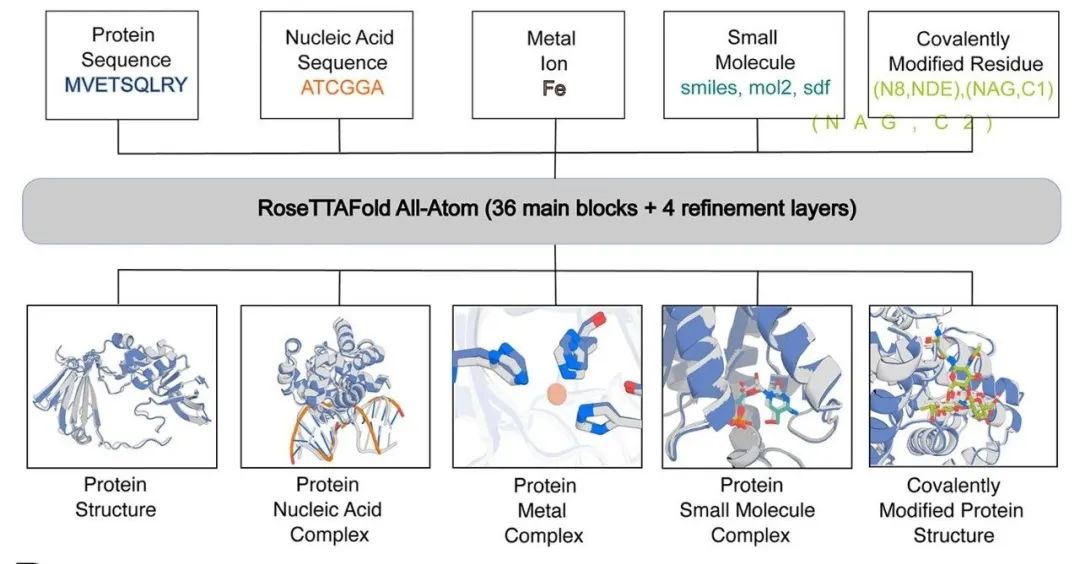
Создание и обучение RFAA
Для проектирования автор использует сеть прогнозирования RoseTTAFold2 (RF2). Представление белков и нуклеиновых кислот в RF2 сохраняется, а малые молекулы, ковалентно модифицированные и неприродные аминокислоты представлены в виде графов (атомов-связей). На 1D-дорожке автор вводит тип химического элемента каждого неполимерного атома, на 2D-дорожке вводятся химические связи между атомами, на 3D-дорожке вводится информация о киральности (R/S);
Автор собрал набор данных о комплексе белок-биомолекула из базы данных PDB, включая комплексы белок-малые молекулы, белок-металл и ковалентно модифицированные белковые комплексы. Общие растворители и добавки были отфильтрованы. Чтобы избежать систематической ошибки, авторы выполнили кластеризацию (идентичность последовательностей 30%). В то же время, чтобы помочь нейронной сети изучить общие свойства малых молекул, а не конкретные особенности в данных PDB, автор использует данные о кристаллической структуре малых молекул в Кембриджской структурной базе данных в качестве дополнительного обучающего набора.
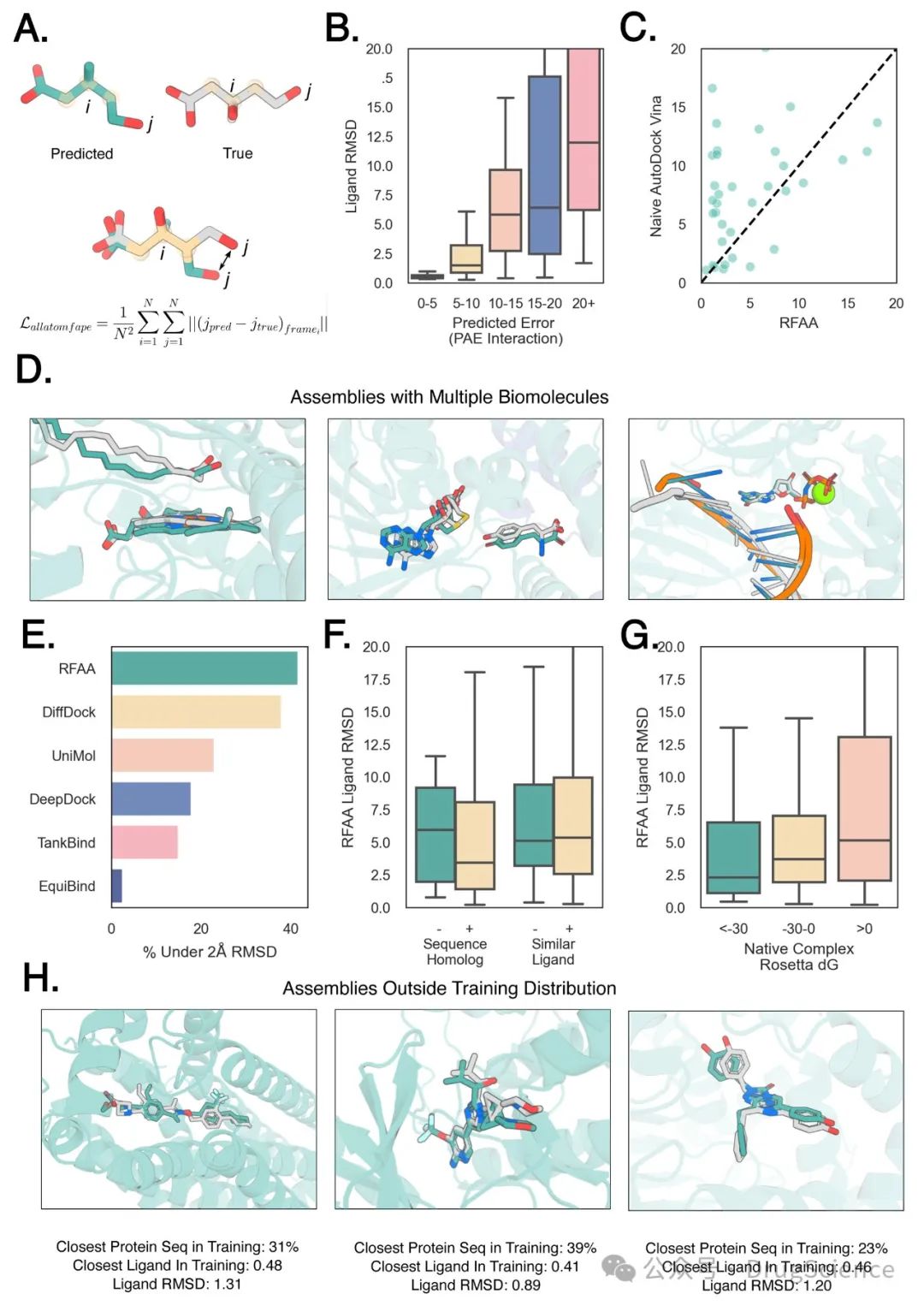
результат
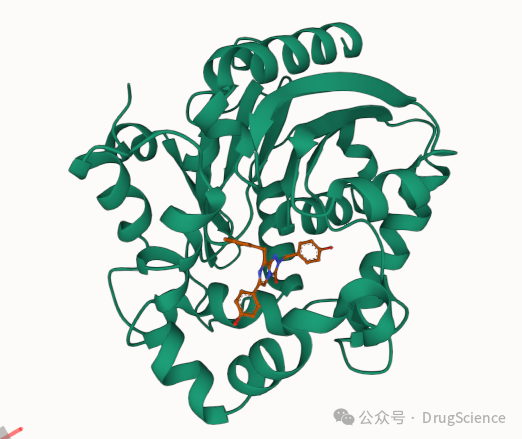
Прогнозирование низкомолекулярных белковых комплексов
Построен сервер RFAA для оценки стыковки CAMEO.,Этот сервер делает еженедельные прогнозы для всех структур, представленных в PDB. В структуре КАМЕО,Есть 43% структура,Результаты прогноза RFAA демонстрируют высокую достоверность (PAE Interaction < 10), среди которых 77% структур с высокой степенью достоверности являются достаточно точными, а RMSD лиганда составляет менее 2 Å, как показано на рисунке выше (B). При этом в сравнении с Виной вероятность успешности предсказания RFAA составила 32%, тогда как Вина — всего 8%. Самая распространенная ошибка RFAA заключается в том, что молекула находится в правильном кармане, но молекула ориентирована неправильно. В то же время следует также отметить, что, если им будут управлять специалисты, возможно, вероятность успешной стыковки Вины будет выше.
Автор также сравнил прогнозы DiffDock и результат RFAA.,Уровень успеха RFAA составляет 42%,Уровень успеха DiffDock составляет 38%.,Но стоит отметить, что RFAA также предсказывает боковые цепи и скелеты белков.,DiffDock напрямую получает структуру белка для стыковки.,RFAA сложнее в эксплуатации.
В случаях, когда предоставляется комбинированная структура белка, Vina работает лучше, чем RFAA (52% против 42%), поскольку перед RFAA стоит более сложная задача по предсказанию деталей основной цепи и боковой цепи белка по последовательности.
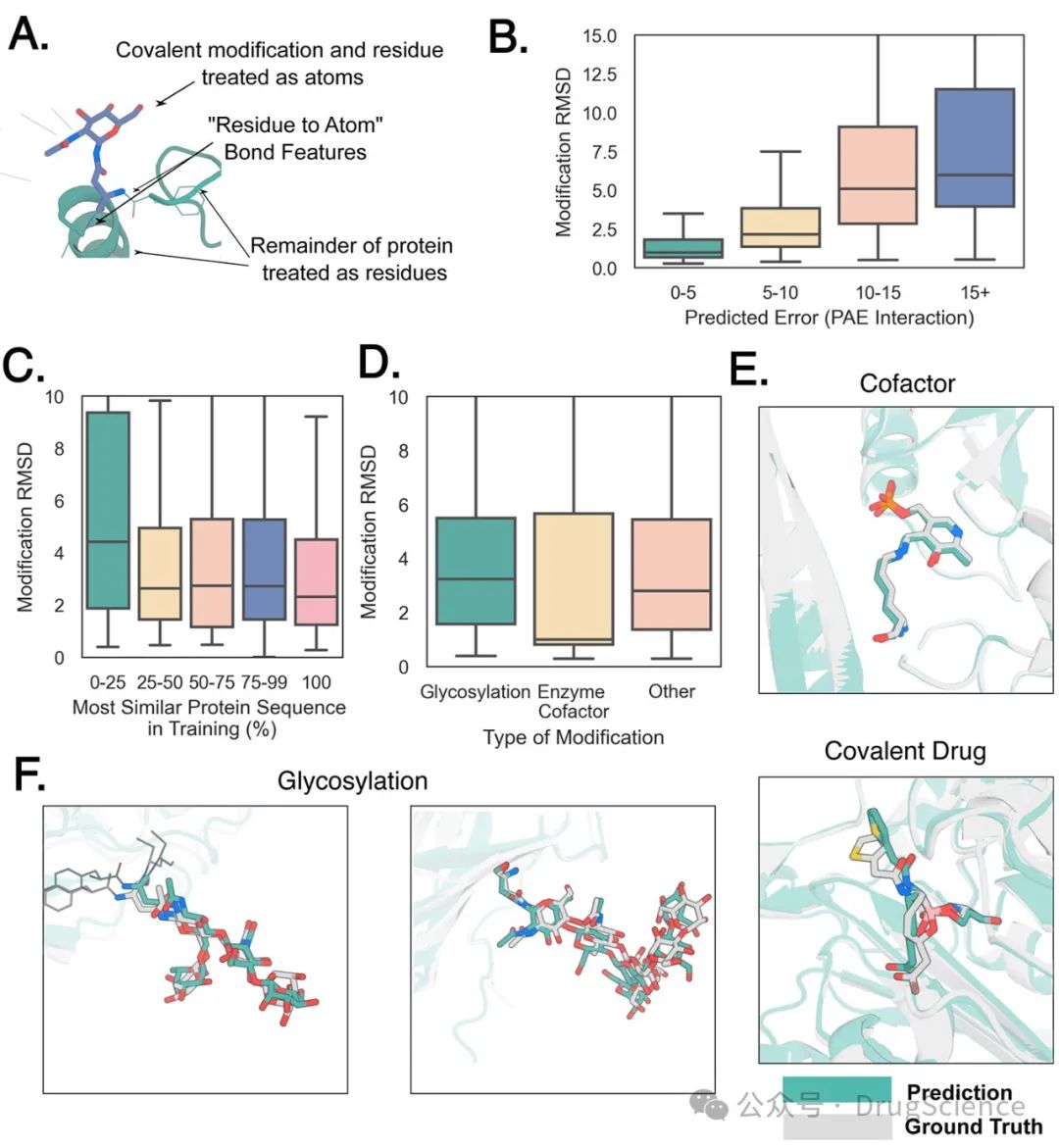
Прогнозирование ковалентных модификаций белка
Авторы предсказали 931 случай ковалентной модификации в данных PDB с вероятностью успеха 46% (RMSD < 2.5Å, Здесь RMSD относится к RMSD модифицированного остатка, когда остальные выровнены. ). В то же время RFAA может эффективно предсказывать структуры гликанов и моделировать углеводные группы, участвующие в гликозилировании. Медиана RMSD на тестовом наборе составляет 3,2. О. RFAA не просто изучает структуру смоделированных гликанов.,Потому что предсказанный результат соответствует экспериментальной карте плотности. Эта сеть работает даже тогда, когда последовательности далеки от тех, что есть в обучающем наборе.,Также позволяет точно прогнозировать взаимодействие гликанов.,И он может предсказывать гликаны, содержащие до семи моносахаридов.
Разработка низкомолекулярных связующих с нуля
В предыдущих исследовательских работах по созданию белков, связывающих небольшие молекулы, обычно использовался метод стыковки молекулы с естественной или выбранной экспертами структурной библиотекой белкового каркаса. Методы, основанные на диффузии, позволяют генерировать белки, которые связываются с белками-мишенями с относительно высоким сродством и специфичностью. С помощью диффузионной обработки авторы могут генерировать белки, которые прочно связываются с небольшими молекулами. На тестовом наборе сгенерированная структура белка очень похожа на реальную структуру белка.
обсуждать
RFAA демонстрирует, что одну нейронную сеть можно обучить точному моделированию биомолекулярных ансамблей, содержащих множество небелковых компонентов. РФАА узнало подробные характеристики белково-малых молекулярных комплексов. Во-первых, сеть способна с высокой точностью предсказывать комплексы, которые существенно отличаются от белков и лигандов в обучающем наборе данных, а энергия взаимодействия, рассчитанная с помощью физической модели Rosetta, помогает повысить точность комплексного предсказания. Во-вторых, RFdiffusionAA также помогает создавать новые белки, объединяющие небольшие молекулы.
Установить
Общая оценка: Установить относительно просто.
Вы можете следовать руководству прямо по этой ссылке: https://github.com/baker-laboratory/RoseTTAFold-All-Atom.
Здесь мы даем некоторые, Введение и меры предосторожности.
1. УстановитьMamba
wget "https://github.com/conda-forge/miniforge/releases/latest/download/Mambaforge-$(uname)-$(uname -m).sh"
bash Mambaforge-$(uname)-$(uname -m).sh # Принять все элементы, восстановить в папку по умолчанию
rm Mambaforge-$(uname)-$(uname -m).sh # (необязательный) Удалить после установки
source ~/.bashrcЗдесь стоит отметить, что если conda существует в вашей исходной среде, исходный путь среды conda будет заменен путем conda mamba.
2. Клонируйте этот пакет.
git clone https://github.com/baker-laboratory/RoseTTAFold-All-Atom
cd RoseTTAFold-All-Atom 3. Используйте мамбу для создания среды RFAA
mamba env create -f environment.yamlconda activate RFAA # NOTE: Это естественно бросить conda deactivate RFAA cd rf2aa/SE3Transformer/pip3 install --no-cache-dir -r requirements.txtpython3 setup.py installcd ../../Операция в основном такая же, как и conda, и никаких других мер предосторожности нет.
4. Загрузите signalp6 и настройте его.
Вы можете выполнить команду «Восстановить» из,зарегистрироваться Академическая версия вашего аккаунта:https://services.healthtech.dtu.dk/services/SignalP-6.0/
Вы получите электронное письмо в свой почтовый ящик и воспользуетесь wget для его загрузки. signalp-6.0h.fast.tar.gz
signalp6-register signalp-6.0h.fast.tar.gz # NOTE: зарегистрироваться
mv $CONDA_PREFIX/lib/python3.10/site-packages/signalp/model_weights/distilled_model_signalp6.pt $CONDA_PREFIX/lib/python3.10/site-packages/signalp/model_weights/ensemble_model_signalp6.pt # ПРИМЕЧАНИЕ. Переименовать distilled Модельмасса 5. Установить Зависимости
bash install_dependencies.sh 6. Загрузите файл веса.
wget http://files.ipd.uw.edu/pub/RF-All-Atom/weights/RFAA_paper_weights.pt 7. Загрузите библиотеку последовательностей, созданную MSA, и шаблон.
# uniref30 [46G]
wget http://wwwuser.gwdg.de/~compbiol/uniclust/2020_06/UniRef30_2020_06_hhsuite.tar.gz
mkdir -p UniRef30_2020_06
tar xfz UniRef30_2020_06_hhsuite.tar.gz -C ./UniRef30_2020_06
# BFD [272G]
wget https://bfd.mmseqs.com/bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt.tar.gz
mkdir -p bfd
tar xfz bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt.tar.gz -C ./bfd
# structure templates (including *_a3m.ffdata, *_a3m.ffindex)
wget https://files.ipd.uw.edu/pub/RoseTTAFold/pdb100_2021Mar03.tar.gz
tar xfz pdb100_2021Mar03.tar.gzЕсли вы раньше играли в AF2,
- Итак, БФДбаза данные не нужно загружать повторно.
- Обратите внимание на версию uniref30базы данных.,Это 06, 2020,Конечно, вы также можете изменить скрипт запуска.,Будет размещен сзади.
- pdb100_2021Mar03.tar.gz,Для загрузки требуется один сеанс<Годовое собрание не может быть остановлено>продолжительность фильма。Возможно, вам нужно распаковать20-30minsо。
8. Необязательно:
- Волябаза данные, создать мягкую ссылку локально
если ты в
RoseTTAFold-All-AtomСкачать и распаковать в папку,Этот шаг можно пропустить. Поскольку файлы моей базы данных хранятся в фиксированном месте.,Поэтому установлено множество мягких ссылок. - Дайте разрешение на создание.sh chmod 777 input_prep/make.sh
бегать
После выполнения вышеизложенного вы можете приступить непосредственно.
# Прогнозирование мономерного белка
python -m rf2aa.run_inference --config-name protein
# Прогнозирование комплекса белково-нуклеиновых кислот
python -m rf2aa.run_inference --config-name nucleic_acid
# Прогнозирование комплекса малых молекул белка
python -m rf2aa.run_inference --config-name protein_smПроцесс запуска занимает около 10-30 минут.
- Прогнозирование структуры белка
- Прогнозирование комплекса малых молекул белка
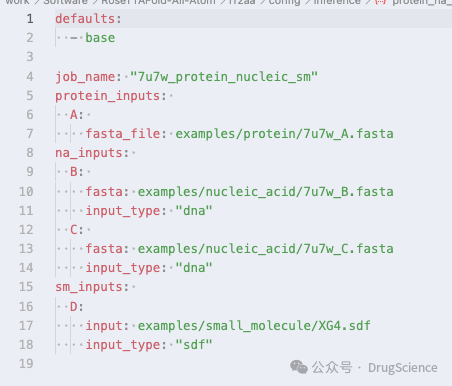
ты можешьrf2aa/config/inferenceКонфигурационный файл находится в,и выполнить индивидуальную настройку.
Ниже особо объяснять нечего.
ссылка
- Rohith Krishna et al.,Generalized biomolecular modeling and design with RoseTTAFold All-Atom.Science0,eadl2528DOI:10.1126/science.adl2528



Неразрушающее увеличение изображений одним щелчком мыши, чтобы сделать их более четкими артефактами искусственного интеллекта, включая руководства по установке и использованию.
Копикодер: этот инструмент отлично работает с Cursor, Bolt и V0! Предоставьте более качественные подсказки для разработки интерфейса (создание навигационного веб-сайта с использованием искусственного интеллекта).
Новый бесплатный RooCline превосходит Cline v3.1? ! Быстрее, умнее и лучше вилка Cline! (Независимое программирование AI, порог 0)

Разработав более 10 проектов с помощью Cursor, я собрал 10 примеров и 60 подсказок.
Я потратил 72 часа на изучение курсорных агентов, и вот неоспоримые факты, которыми я должен поделиться!

Идеальная интеграция Cursor и DeepSeek API
DeepSeek V3 снижает затраты на обучение больших моделей
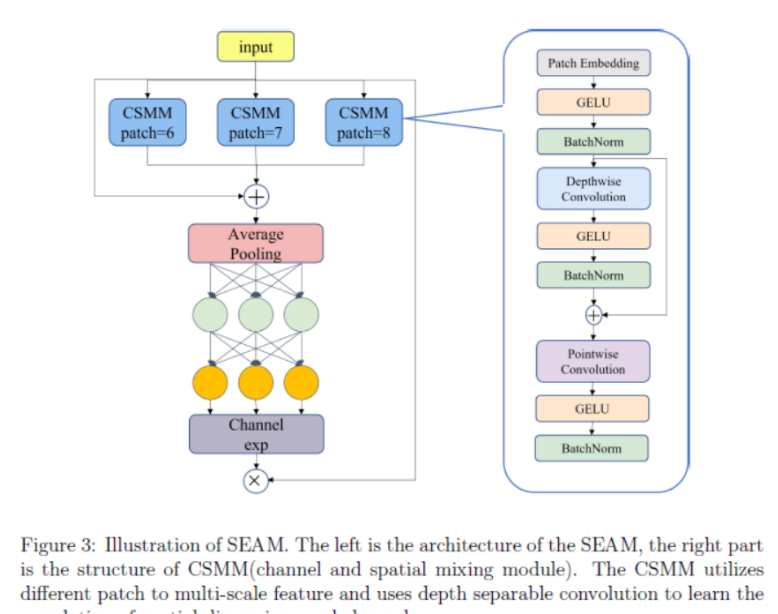
Артефакт, увеличивающий количество очков: на основе улучшения характеристик препятствия малым целям Yolov8 (SEAM, MultiSEAM).

DeepSeek V3 раскручивался уже три дня. Сегодня я попробовал самопровозглашенную модель «ChatGPT».

Open Devin — инженер-программист искусственного интеллекта с открытым исходным кодом, который меньше программирует и больше создает.
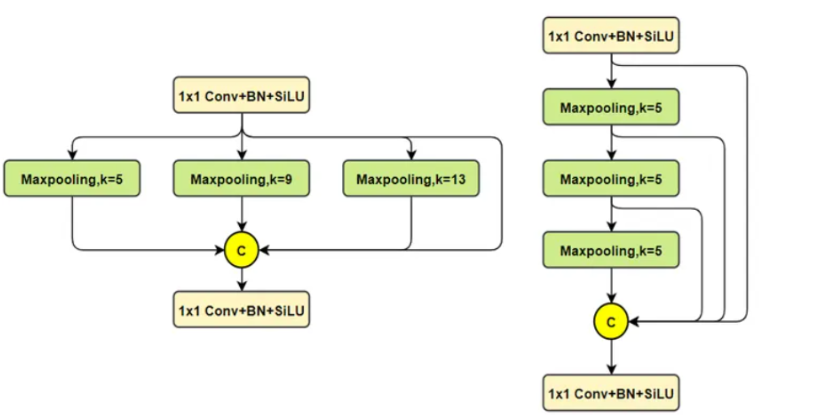
Эксклюзивное оригинальное улучшение YOLOv8: собственная разработка SPPF | SPPF сочетается с воспринимаемой большой сверткой ядра UniRepLK, а свертка с большим ядром + без расширения улучшает восприимчивое поле

Популярное и подробное объяснение DeepSeek-V3: от его появления до преимуществ и сравнения с GPT-4o.

9 основных словесных инструкций по доработке академических работ с помощью ChatGPT, эффективных и практичных, которые стоит собрать
Вызовите deepseek в vscode для реализации программирования с помощью искусственного интеллекта.
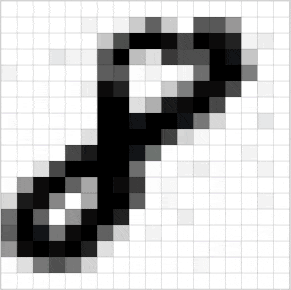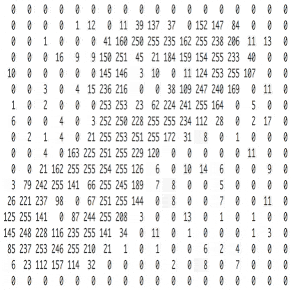
Познакомьтесь с принципами сверточных нейронных сетей (CNN) в одной статье (суперподробно)
50,3 тыс. звезд! Immich: автономное решение для резервного копирования фотографий и видео, которое экономит деньги и избавляет от беспокойства.
Cloud Native|Практика: установка Dashbaord для K8s, графика неплохая
Краткий обзор статьи — использование синтетических данных при обучении больших моделей и оптимизации производительности
MiniPerplx: новая поисковая система искусственного интеллекта с открытым исходным кодом, спонсируемая xAI и Vercel.
Конструкция сервиса Synology Drive сочетает проникновение в интрасеть и синхронизацию папок заметок Obsidian в облаке.
Центр конфигурации————Накос
Начинаем с нуля при разработке в облаке Copilot: начать разработку с минимальным использованием кода стало проще

[Серия Docker] Docker создает мультиплатформенные образы: практика архитектуры Arm64

Обновление новых возможностей coze | Я использовал coze для создания апплета помощника по исправлению домашних заданий по математике

Советы по развертыванию Nginx: практическое создание статических веб-сайтов на облачных серверах
Feiniu fnos использует Docker для развертывания личного блокнота Notepad

Сверточная нейронная сеть VGG реализует классификацию изображений Cifar10 — практический опыт Pytorch
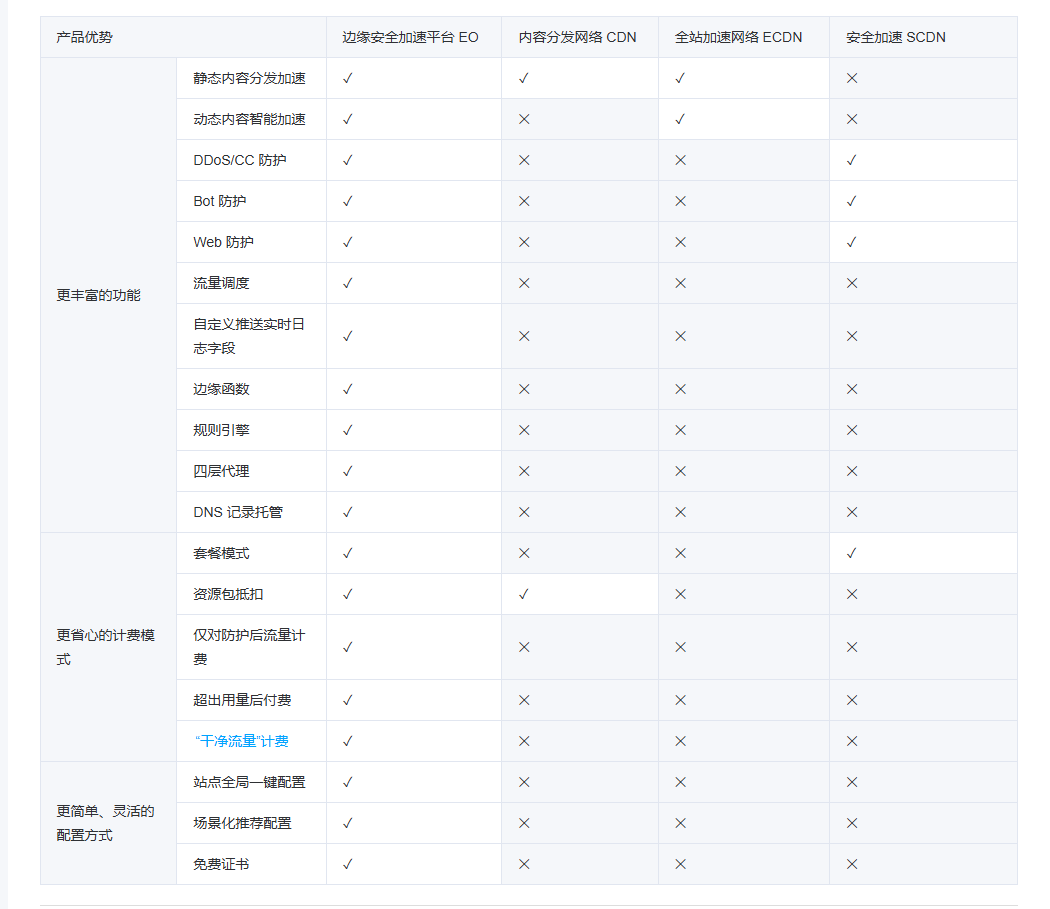
Начало работы с EdgeonePages — новым недорогим решением для хостинга веб-сайтов
[Зона легкого облачного игрового сервера] Управление игровыми архивами