irGSEA: интегрированная система для рангового анализа обогащения набора генов одной клетки
фон
Много Функционала Class Scoring (FCS)метод,нравитьсяGSEA, GSVA,PLAGE, addModuleScore, SCSE, Vision, VAM, gficf, pagoda2иSargent,будет зависеть от состава набора данных из,Небольшие изменения в составе набора данных изменят показатель обогащения набора генов.
Если новый набор данных из одной ячейки интегрирован в существующие данные,используйте этиFCSметод需要重新计算每个细胞изпоказатель обогащения генного набора。Этот шаг может быть утомительным и ресурсоемким.。
Напротив,На основе уровня экспрессии отдельных клеток изFCS,нравитьсяAUCell、UCell、singscore、ssGSEA、JASMINEиViper,Необходимо только рассчитать показатель обогащения вновь добавленных наборов одноячеечных данных из,Без пересчета показателей обогащения всех клеток из набора генов. Причина в том, что эти методы генерируют показатели обогащения, которые полагаются только на относительную экспрессию генов на уровне отдельных клеток.,Ничего общего с составом набора данных. поэтому,Эти методы могут сэкономить много времени.
просмотреть результаты
здесь,мы рассмотрели17 распространенных методов FCS:
GSEAОбнаружение степени обогащения наборов генов вверху или внизу отсортированного списка генов. Список получается путем расчета отношения сигнал-шум отсортированных генов или кратного изменения отсортированных генов после группировки;GSVAОцените ядро функции совокупной плотности для каждого гена во всех клетках. В этом процессе необходимо учитывать все образцы, и информация об образцах легко влияет на них;PLAGEНормализовать матрицу экспрессии генов в клетках и извлечь разложение по сингулярным значениям в виде показателя обогащения набора генов;ZscoreАгрегирует экспрессию всех генов в наборе генов, масштабируя экспрессию по среднему значению и стандартному отклонению по клеткам;AddModuleScoreВсе гены в наборе генов необходимо сначала рассчитать.изсреднее значение,Затем разрежьте матрицу выражений на несколько частей на основе среднего значения,Затем из каждого среза после среза в качестве значения фон случайным образом выбираются контрольные гены (гены вне набора генов). поэтому,В случае объединения разных выборок,Даже если одна и та же клетка забита с использованием одного и того же набора генов,Также будут генерироваться различные оценки обогащения;SCSEПоказатели обогащения набора генов количественно оцениваются с использованием нормализованной суммы всех генов в наборе генов;VisionОценки обогащения набора генов определялись с использованием ожидаемого среднего значения и дисперсии случайных сигнатур. z Нормализовать, чтобы исправить показатели обогащения набора генов;VAMСогласно классическому многомерному расстоянию Махаланобиса от одиночных клеток RNA Данные секвенирования позволяют получить показатели обогащения набора генов;GficfИнформационные биологические сигналы, использующие латентные факторы значений экспрессии генов, полученные посредством факторизации неотрицательной матрицы;Pagoda2Подберите модель ошибок для каждой клетки и количественно оцените показатель обогащения набора генов, используя его первый взвешенный главный компонент;AUCellНа основе ранжирования экспрессии генов в одном образце используйте область под кривой, чтобы оценить, обогащен ли входной набор генов 5% наиболее экспрессируемых генов в одном образце;UCellРанжирование экспрессии генов на основе отдельных образцов с использованием метода Манна-Уитни. Статистика U рассчитывает показатель обогащения набора генов для одного образца;SingscoreРасстояние от центра отдельных клеток оценивали на основе уровней экспрессии генов. Гены в наборе генов упорядочены на основе количества транскриптов в отдельных клетках. Средний ранг индивидуально нормализуется относительно теоретических минимального и максимального значений, центрируется вокруг нуля, а затем суммируется, и полученная оценка представляет собой оценку обогащения набора генов;ssGSEAПоказатель разницы эмпирического кумулятивного распределения между внутренним и внешним наборами генов рассчитывали на основе ранга экспрессии генов в каждой клетке. Оценки различий были нормализованы с использованием глобальных профилей экспрессии. На этот этап стандартизации легко влияет состав образца.JASMINEПриблизительные средние значения рассчитываются на основе ранжирования генов среди генов, экспрессируемых в отдельных клетках, и обогащения наборов генов среди экспрессируемых генов. Оба значения нормированы на 0-1 диапазонов и объединяются путем усреднения для получения окончательного показателя обогащения набора генов.ViperПоказатель обогащения набора генов оценивается путем выполнения трехстороннего расчета, основанного на ранжировании экспрессии генов в клетках.SargentСортируйте ненулевые экспрессированные гены для данной клетки от высокой экспрессии до низкой экспрессии и преобразуйте входную матрицу экспрессии генов в клетках в соответствующий набор генов для каждой клетки. assignment score matrix。 但SargentПосле того, как вам понадобится рассчитать индекс Джини между клетками, нажмите «Набор генов по ячейкам». assignment score матрица, преобразованная в распределение of indexes。
Рабочий процесс
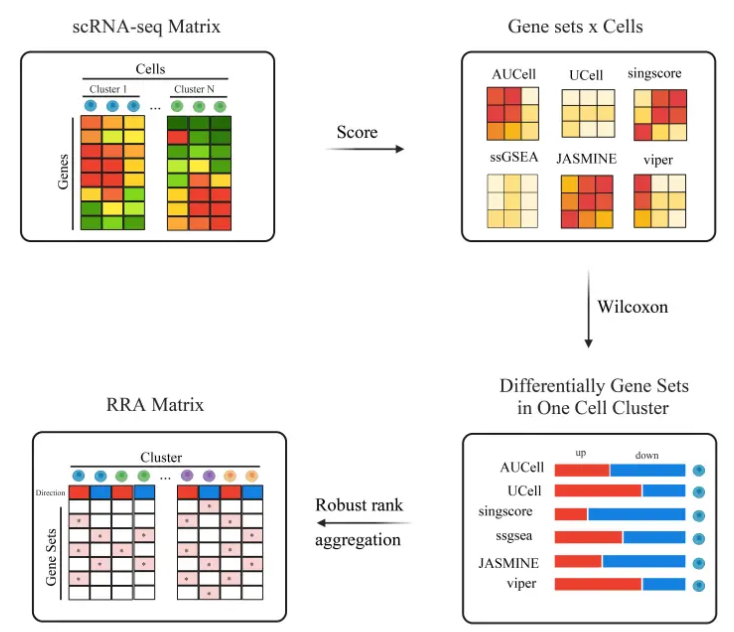
- использовать
AUCell、UCell、singscore、ssgsea、JASMINE и viperОцените каждую ячейку отдельно,Получите различные матрицы оценок обогащения. - проходить
тест УилкоксонаРасчет другойиз Дифференциальная экспрессия каждой субпопуляции клеток в матрице показателей обогащенияизгенный набор。вверх или вниз表示该细胞簇内差异генный набориз Обогащен более или менее, чем другие кластеры。
Метод анализа обогащения одного генного набора может не только отражать ограниченную информацию.,Также легко вызвать ошибки。Мы стремимся объяснить сложные вопросы с разных точек зрения.избиологические вопросы,И найти общее в биологических задачах. Просто возьмите общее пересечение результатов нескольких методов анализа обогащения набора генов.,Мало того, что легко получить меньше, но и консервативные результаты,И он игнорирует много другой информации в методе анализа обогащения.,Например, информация об относительном обогащении разных наборов генов.
Мы ожидаем, что целевой набор генов будет обогащен большинством методов анализа обогащения и что не будет существенной разницы в степени обогащения.。поэтому,我们проходитьRobustRankAggregВ сумкеизНадежное агрегирование рангов (RRA)Анализ различийиз Оцените результаты,Отобраны наборы генов, которые показали одинаковые уровни обогащения и различия в генах среди 6 методов.
установка irGSEA
1.установка irGSEA (базовая конфигурация)
Использует только AUCell, UCell, Singscore, ssGSEA, JASMINE и viper.
# install packages from CRAN
cran.packages <- c("aplot", "BiocManager", "data.table", "devtools",
"doParallel", "doRNG", "dplyr", "ggfun", "gghalves",
"ggplot2", "ggplotify", "ggridges", "ggsci", "irlba",
"magrittr", "Matrix", "msigdbr", "pagoda2", "pointr",
"purrr", "RcppML", "readr", "reshape2", "reticulate",
"rlang", "RMTstat", "RobustRankAggreg", "roxygen2",
"Seurat", "SeuratObject", "stringr", "tibble", "tidyr",
"tidyselect", "tidytree", "VAM")
for (i in cran.packages) {
if (!requireNamespace(i, quietly = TRUE)) {
install.packages(i, ask = F, update = F)
}
}
# install packages from Bioconductor
bioconductor.packages <- c("AUCell", "BiocParallel", "ComplexHeatmap",
"decoupleR", "fgsea", "ggtree", "GSEABase",
"GSVA", "Nebulosa", "scde", "singscore",
"SummarizedExperiment", "UCell",
"viper","sparseMatrixStats")
for (i in bioconductor.packages) {
if (!requireNamespace(i, quietly = TRUE)) {
install.packages(i, ask = F, update = F)
}
}
# install packages from Github
if (!requireNamespace("irGSEA", quietly = TRUE)) {
devtools::install_github("chuiqin/irGSEA", force =T)
}
2.установка irGSEA (расширенная конфигурация)
Хотите использовать VISION, gficf, Sargent, ssGSEApy, GSVApy и другие методы (эта часть является дополнительной установкой)
# VISION
if (!requireNamespace("VISION", quietly = TRUE)) {
devtools::install_github("YosefLab/VISION", force =T)
}
# mdt need ranger
if (!requireNamespace("ranger", quietly = TRUE)) {
devtools::install_github("imbs-hl/ranger", force =T)
}
# gficf need RcppML (version > 0.3.7) package
if (!utils::packageVersion("RcppML") > "0.3.7") {
message("The version of RcppML should greater than 0.3.7 and install RcppML package from Github")
devtools::install_github("zdebruine/RcppML", force =T)
}
# please first `library(RcppML)` if you want to perform gficf
if (!requireNamespace("gficf", quietly = TRUE)) {
devtools::install_github("gambalab/gficf", force =T)
}
# GSVApy and ssGSEApy need SeuratDisk package
if (!requireNamespace("SeuratDisk", quietly = TRUE)) {
devtools::install_github("mojaveazure/seurat-disk", force =T)
}
# sargent
if (!requireNamespace("sargent", quietly = TRUE)) {
devtools::install_github("Sanofi-Public/PMCB-Sargent", force =T)
}
# pagoda2 need scde package
if (!requireNamespace("scde", quietly = TRUE)) {
devtools::install_github("hms-dbmi/scde", force =T)
}
# if error1 (functio 'sexp_as_cholmod_sparse' not provided by package 'Matrix')
# or error2 (functio 'as_cholmod_sparse' not provided by package 'Matrix') occurs
# when you perform pagoda2, please check the version of irlba and Matrix
# It's ok when I test as follow:
# R 4.2.2 irlba(v 2.3.5.1) Matrix(1.5-3)
# R 4.3.1 irlba(v 2.3.5.1) Matrix(1.6-1.1)
# R 4.3.2 irlba(v 2.3.5.1) Matrix(1.6-3)
#### create conda env
# If error (Unable to find conda binary. Is Anaconda installed) occurs,
# please perform `reticulate::install_miniconda()`
if (! "irGSEA" %in% reticulate::conda_list()$name) {
reticulate::conda_create("irGSEA")
}
# if python package exist
python.package <- reticulate::py_list_packages(envname = "irGSEA")$package
require.package <- c("anndata", "scanpy", "argparse", "gseapy", "decoupler")
for (i in seq_along(require.package)) {
if (i %in% python.package) {
reticulate::conda_install(envname = "irGSEA", packages = i, pip = T)
}
}
3. Ускоренная установка внутреннего образа
Если у вас возникли трудности с установкой пакетов github и pip, обратитесь сюда. Я клонировал пакет github в gitee.
options(BioC_mirror="https://mirrors.tuna.tsinghua.edu.cn/bioconductor/")
options("repos" = c(CRAN="http://mirrors.cloud.tencent.com/CRAN/"))
# install packages from CRAN
cran.packages <- c("aplot", "BiocManager", "data.table", "devtools",
"doParallel", "doRNG", "dplyr", "ggfun", "gghalves",
"ggplot2", "ggplotify", "ggridges", "ggsci", "irlba",
"magrittr", "Matrix", "msigdbr", "pagoda2", "pointr",
"purrr", "RcppML", "readr", "reshape2", "reticulate",
"rlang", "RMTstat", "RobustRankAggreg", "roxygen2",
"Seurat", "SeuratObject", "stringr", "tibble", "tidyr",
"tidyselect", "tidytree", "VAM")
for (i in cran.packages) {
if (!requireNamespace(i, quietly = TRUE)) {
install.packages(i, ask = F, update = F)
}
}
# install packages from Bioconductor
bioconductor.packages <- c("AUCell", "BiocParallel", "ComplexHeatmap",
"decoupleR", "fgsea", "ggtree", "GSEABase",
"GSVA", "Nebulosa", "scde", "singscore",
"SummarizedExperiment", "UCell", "viper")
for (i in bioconductor.packages) {
if (!requireNamespace(i, quietly = TRUE)) {
install.packages(i, ask = F, update = F)
}
}
# install packages from git
if (!requireNamespace("irGSEA", quietly = TRUE)) {
devtools::install_git("https://gitee.com/fan_chuiqin/irGSEA.git", force =T)
}
# VISION
if (!requireNamespace("VISION", quietly = TRUE)) {
devtools::install_git("https://gitee.com/fan_chuiqin/VISION.git", force =T)
}
# mdt need ranger
if (!requireNamespace("ranger", quietly = TRUE)) {
devtools::install_git("https://gitee.com/fan_chuiqin/ranger.git", force =T)
}
# gficf need RcppML (version > 0.3.7) package
if (!utils::packageVersion("RcppML") > "0.3.7") {
message("The version of RcppML should greater than 0.3.7 and install RcppML package from Git")
devtools::install_git("https://gitee.com/fan_chuiqin/RcppML.git", force =T)
}
# please first `library(RcppML)` if you want to perform gficf
if (!requireNamespace("gficf", quietly = TRUE)) {
devtools::install_git("https://gitee.com/fan_chuiqin/gficf.git", force =T)
}
# GSVApy and ssGSEApy need SeuratDisk package
if (!requireNamespace("SeuratDisk", quietly = TRUE)) {
devtools::install_git("https://gitee.com/fan_chuiqin/seurat-disk.git",
force =T)}
# sargent
if (!requireNamespace("sargent", quietly = TRUE)) {
devtools::install_git("https://gitee.com/fan_chuiqin/PMCB-Sargent.git",
force =T)}
# pagoda2 need scde package
if (!requireNamespace("scde", quietly = TRUE)) {
devtools::install_git("https://gitee.com/fan_chuiqin/scde.git", force =T)
}
#### create conda env
# If error (Unable to find conda binary. Is Anaconda installed) occurs,
# please perform `reticulate::install_miniconda()`
if (! "irGSEA" %in% reticulate::conda_list()$name) {
reticulate::conda_create("irGSEA")
}
# if python package exist
python.package <- reticulate::py_list_packages(envname = "irGSEA")$package
require.package <- c("anndata", "scanpy", "argparse", "gseapy", "decoupler")
for (i in require.package) {
if (! i %in% python.package) {
reticulate::conda_install(envname = "irGSEA", packages = i, pip = T,
pip_options = "-i https://pypi.tuna.tsinghua.edu.cn/simple")
}
}
Учебное пособие
1.irGSEA поддерживает объект Seurat (V5 или V4), объект анализа (V5 или V4).
# Мы загружаем пример набора данных (хорошо аннотированный из набора данных PBMC) через пакет SeuratData в качестве демонстрации.
#### Seurat V4объект ####
library(Seurat)
library(SeuratData)
library(RcppML)
library(irGSEA)
data("pbmc3k.final")
pbmc3k.final <- irGSEA.score(object = pbmc3k.final,assay = "RNA",
slot = "data", seeds = 123, ncores = 1,
min.cells = 3, min.feature = 0,
custom = F, geneset = NULL, msigdb = T,
species = "Homo sapiens", category = "H",
subcategory = NULL, geneid = "symbol",
method = c("AUCell","UCell","singscore","ssgsea", "JASMINE", "viper"),
aucell.MaxRank = NULL, ucell.MaxRank = NULL,
kcdf = 'Gaussian')
Assays(pbmc3k.final)
>[1] "RNA" "AUCell" "UCell" "singscore" "ssgsea" "JASMINE" "viper"
#### Seurat V5объект ####
data("pbmc3k.final")
pbmc3k.final <- SeuratObject::UpdateSeuratObject(pbmc3k.final)
pbmc3k.final2 <- CreateSeuratObject(counts = CreateAssay5Object(GetAssayData(pbmc3k.final, assay = "RNA", slot = "counts")),
meta.data = pbmc3k.final[[]])
pbmc3k.final2 <- NormalizeData(pbmc3k.final2)
pbmc3k.final2 <- irGSEA.score(object = pbmc3k.final2,assay = "RNA",
slot = "data", seeds = 123, ncores = 1,
min.cells = 3, min.feature = 0,
custom = F, geneset = NULL, msigdb = T,
species = "Homo sapiens", category = "H",
subcategory = NULL, geneid = "symbol",
method = c("AUCell","UCell","singscore","ssgsea", "JASMINE", "viper"),
aucell.MaxRank = NULL, ucell.MaxRank = NULL,
kcdf = 'Gaussian')
Assays(pbmc3k.final2)
[1] "RNA" "AUCell" "UCell" "singscore" "ssgsea" "JASMINE" "viper"
#### Assay5 объект ####
data("pbmc3k.final")
pbmc3k.final <- SeuratObject::UpdateSeuratObject(pbmc3k.final)
pbmc3k.final3 <- CreateAssay5Object(counts = GetAssayData(pbmc3k.final, assay = "RNA", slot = "counts"))
pbmc3k.final3 <- NormalizeData(pbmc3k.final3)
pbmc3k.final3 <- irGSEA.score(object = pbmc3k.final3,assay = "RNA",
slot = "counts", seeds = 123, ncores = 1,
min.cells = 3, min.feature = 0,
custom = F, geneset = NULL, msigdb = T,
species = "Homo sapiens", category = "H",
subcategory = NULL, geneid = "symbol",
method = c("AUCell","UCell","singscore","ssgsea", "JASMINE", "viper"),
aucell.MaxRank = NULL, ucell.MaxRank = NULL,
kcdf = 'Gaussian')
Assays(pbmc3k.final3)
# Assay5объект хранится в РНК Во время анализа
Статистика размера файла результатов:
# Смотри, Сёра V5иAssay 5 Это правда, что объем хранения данных будет относительно небольшим.
> cat(object.size(pbmc3k.final) / (1024^2), "M")
339.955 M
> cat(object.size(pbmc3k.final2) / (1024^2), "M")
69.33521 M
> cat(object.size(pbmc3k.final3) / (1024^2), "M")
69.27851 M
2. Методы оценки набора генов, поддерживаемые irGSEA.
Мы протестировали пик времени и памяти, необходимый для оценки, используя 50 наборов генов Hallmark для различных методов оценки при разных размерах данных. Вы можете выбирать в соответствии с вашим собственным компьютером и временем;

GSVApy, ssGSEApy и viperpy представляют собой версии GSVA, ssGSEA и viper для Python соответственно. Версия GSVA для Python экономит слишком много времени, чем версия GSVA для R.
Наш рейтинг、ssGSEA、JASMINE、viperиз Пик памяти оптимизирован。 Более чем 50000 ячеек, мы реализовали стратегию разделения их на 5000 клетки/единицы были оценены. Хотя это устраняет проблемы с перегрузками памяти, но увеличивает время обработки.
3. Методы оценки набора генов, поддерживаемые irGSEA.
Чтобы облегчить пользователям получение предопределенных наборов генов в библиотеке MSigDBданные.,Мы встроилипакет msigdbrруководитьMSigDBизгенный наборданныеизполучать。пакет msigdbrПоддерживает несколько видовизгенный наборполучать,а такжеНесколько форматов геновизматрица выраженийизвходить。
①irGSEA оценивается с помощью встроенного пакета msigdbr.
library(Seurat)
library(SeuratData)
library(RcppML)
library(irGSEA)
data("pbmc3k.final")
#### Оценка набора генов Hallmark ####
pbmc3k.final <- irGSEA.score(object = pbmc3k.final,assay = "RNA",
slot = "data", seeds = 123, ncores = 1,
min.cells = 3, min.feature = 0,
custom = F, geneset = NULL, msigdb = T,
species = "Homo sapiens", category = "H",
subcategory = NULL, geneid = "symbol",
method = c("AUCell","UCell","singscore","ssgsea", "JASMINE", "viper"),
aucell.MaxRank = NULL, ucell.MaxRank = NULL,
kcdf = 'Gaussian')
#### Оценка набора генов KEGG ####
pbmc3k.final <- irGSEA.score(object = pbmc3k.final,assay = "RNA",
slot = "data", seeds = 123, ncores = 1,
min.cells = 3, min.feature = 0,
custom = F, geneset = NULL, msigdb = T,
species = "Homo sapiens", category = "C2",
subcategory = "CP:KEGG", geneid = "symbol",
method = c("AUCell","UCell","singscore","ssgsea", "JASMINE", "viper"),
aucell.MaxRank = NULL, ucell.MaxRank = NULL,
kcdf = 'Gaussian')
#### Оценка набора генов GO-BP ####
pbmc3k.final <- irGSEA.score(object = pbmc3k.final,assay = "RNA",
slot = "data", seeds = 123, ncores = 1,
min.cells = 3, min.feature = 0,
custom = F, geneset = NULL, msigdb = T,
species = "Homo sapiens", category = "C5",
subcategory = "GO:BP", geneid = "symbol",
method = c("AUCell","UCell","singscore","ssgsea", "JASMINE", "viper"),
aucell.MaxRank = NULL, ucell.MaxRank = NULL,
kcdf = 'Gaussian')
②irGSEA использует последнюю версию набора генов MSigDB для оценки.
Сожаление,
пакет msigdbrвстроенныйизMSigDBизверсияMSigDB 2022.1。Однако,в настоящий моментMSigDBданные Библиотека обновлена до2023.2,включать2023.2.Hsи2023.2.Mm。Мы можем использовать эту ссылку(https://data.broadinstitute.org/gsea-msigdb/msigdb/release/)скачать2023.2.HsизGMT-файлилифайл db.zip。по сравнению сGMT-файл,файл db.zip包含了генный наборизописывать,Его можно использовать для скрининга генов, связанных с функцией XX. В примере ниже,我将介绍нравиться何筛选Набор генов, связанных с ангиогенезом。
#### work with newest Msigdb ####
# https://data.broadinstitute.org/gsea-msigdb/msigdb/release/
# In this page, you can download human/mouse gmt file or db.zip file
# The db.zip file contains metadata information for the gene set
# load library
library(clusterProfiler)
library(tidyverse)
library(DBI)
library(RSQLite)
### db.zip ###
# download zip file and unzip zip file
zip_url <- "https://data.broadinstitute.org/gsea-msigdb/msigdb/release/2023.2.Hs/msigdb_v2023.2.Hs.db.zip"
local_zip_path <- "./msigdb_v2023.2.Hs.db.zip"
download.file(zip_url, local_zip_path)
unzip(local_zip_path, exdir = "./")
# code modified by https://rdrr.io/github/cashoes/sear/src/data-raw/1_parse_msigdb_sqlite.r
con <- DBI::dbConnect(RSQLite::SQLite(), dbname = './msigdb_v2023.2.Hs.db')
DBI::dbListTables(con)
# define tables we want to combine
geneset_db <- dplyr::tbl(con, 'gene_set') # standard_name, collection_name
details_db <- dplyr::tbl(con, 'gene_set_details') # description_brief, description_full
geneset_genesymbol_db <- dplyr::tbl(con, 'gene_set_gene_symbol') # meat and potatoes
genesymbol_db <- dplyr::tbl(con, 'gene_symbol') # mapping from ids to gene symbols
collection_db <- dplyr::tbl(con, 'collection') %>% dplyr::select(collection_name, full_name) # collection metadata
# join tables
msigdb <- geneset_db %>%
dplyr::left_join(details_db, by = c('id' = 'gene_set_id')) %>%
dplyr::left_join(collection_db, by = 'collection_name') %>%
dplyr::left_join(geneset_genesymbol_db, by = c('id' = 'gene_set_id')) %>%
dplyr::left_join(genesymbol_db, by = c('gene_symbol_id' = 'id')) %>%
dplyr::select(collection = collection_name, subcollection = full_name, geneset = standard_name, description = description_brief, symbol) %>%
dplyr::as_tibble()
# clean up
DBI::dbDisconnect(con)
unique(msigdb$collection)
# [1] "C1" "C2:CGP" "C2:CP:BIOCARTA"
# [4] "C2:CP:KEGG_LEGACY" "C2:CP:PID" "C3:MIR:MIRDB"
# [7] "C3:MIR:MIR_LEGACY" "C3:TFT:GTRD" "C3:TFT:TFT_LEGACY"
# [10] "C4:3CA" "C4:CGN" "C4:CM"
# [13] "C6" "C7:IMMUNESIGDB" "C7:VAX"
# [16] "C8" "C5:GO:BP" "C5:GO:CC"
# [19] "C5:GO:MF" "H" "C5:HPO"
# [22] "C2:CP:KEGG_MEDICUS" "C2:CP:REACTOME" "C2:CP:WIKIPATHWAYS"
# [25] "C2:CP"
unique(msigdb$subcollection)
# [1] "C1" "C2:CGP" "C2:CP:BIOCARTA"
# [4] "C2:CP:KEGG_LEGACY" "C2:CP:PID" "C3:MIR:MIRDB"
# [7] "C3:MIR:MIR_LEGACY" "C3:TFT:GTRD" "C3:TFT:TFT_LEGACY"
# [10] "C4:3CA" "C4:CGN" "C4:CM"
# [13] "C6" "C7:IMMUNESIGDB" "C7:VAX"
# [16] "C8" "C5:GO:BP" "C5:GO:CC"
# [19] "C5:GO:MF" "H" "C5:HPO"
# [22] "C2:CP:KEGG_MEDICUS" "C2:CP:REACTOME" "C2:CP:WIKIPATHWAYS"
# [25] "C2:CP"
# convert to list[Hallmark] required by irGSEA package
msigdb.h <- msigdb %>%
dplyr::filter(collection=="H") %>%
dplyr::select(c("geneset", "symbol"))
msigdb.h$geneset <- factor(msigdb.h$geneset)
msigdb.h <- msigdb.h %>%
dplyr::group_split(geneset, .keep = F) %>%
purrr::map( ~.x %>% dplyr::pull(symbol) %>% unique(.)) %>%
purrr::set_names(levels(msigdb.h$geneset))
#### Оценка набора генов Hallmark ####
pbmc3k.final <- irGSEA.score(object = pbmc3k.final, assay = "RNA", slot = "data",
custom = T, geneset = msigdb.h,
method = c("AUCell","UCell","singscore","ssgsea", "JASMINE", "viper"),
kcdf = 'Gaussian')
# convert to list[go bp] required by irGSEA package
msigdb.go.bp <- msigdb %>%
dplyr::filter(collection=="C5:GO:BP") %>%
dplyr::select(c("geneset", "symbol"))
msigdb.go.bp$geneset <- factor(msigdb.go.bp$geneset)
msigdb.go.bp <- msigdb.go.bp %>%
dplyr::group_split(geneset, .keep = F) %>%
purrr::map( ~.x %>% dplyr::pull(symbol) %>% unique(.)) %>%
purrr::set_names(levels(msigdb.go.bp$geneset))
#### Оценка набора генов GO-BP ####
pbmc3k.final <- irGSEA.score(object = pbmc3k.final, assay = "RNA", slot = "data",
custom = T, geneset = msigdb.go.bp,
method = c("AUCell","UCell","singscore","ssgsea", "JASMINE", "viper"),
kcdf = 'Gaussian')
# convert to list[KEGG] required by irGSEA package
msigdb.kegg <- msigdb %>%
dplyr::filter(collection=="C2:CP:KEGG_MEDICUS") %>%
dplyr::select(c("geneset", "symbol"))
msigdb.kegg$geneset <- factor(msigdb.kegg$geneset)
msigdb.kegg <- msigdb.kegg %>%
dplyr::group_split(geneset, .keep = F) %>%
purrr::map( ~.x %>% dplyr::pull(symbol) %>% unique(.)) %>%
purrr::set_names(levels(msigdb.kegg$geneset))
#### Оценка набора генов KEGG ####
pbmc3k.final <- irGSEA.score(object = pbmc3k.final, assay = "RNA", slot = "data",
custom = T, geneset = msigdb.kegg,
method = c("AUCell","UCell","singscore","ssgsea", "JASMINE", "viper"),
kcdf = 'Gaussian')
# Look for the gene sets associated with angiogenesis from gene sets names and
# gene sets descriptions
category <- c("angiogenesis", "vessel")
msigdb.vessel <- list()
for (i in category) {
# Ignore case matching
find.index.description <- stringr::str_detect(msigdb$description, pattern = regex(all_of(i), ignore_case=TRUE))
find.index.name <- stringr::str_detect(msigdb$geneset, pattern = regex(all_of(i), ignore_case=TRUE))
msigdb.vessel[[i]] <- msigdb[find.index.description | find.index.name, ] %>% mutate(category = i)
}
msigdb.vessel <- do.call(rbind, msigdb.vessel)
head(msigdb.vessel)
# # A tibble: 6 × 6
# collection subcollection geneset description symbol category
# <chr> <chr> <chr> <chr> <chr> <chr>
# 1 C2:CGP Chemical and Genetic Perturbations HU_ANGIOGENESIS_UP Up-regulated … HECW1 angioge…
# 2 C2:CGP Chemical and Genetic Perturbations HU_ANGIOGENESIS_UP Up-regulated … JADE2 angioge…
# 3 C2:CGP Chemical and Genetic Perturbations HU_ANGIOGENESIS_UP Up-regulated … SEMA3C angioge…
# 4 C2:CGP Chemical and Genetic Perturbations HU_ANGIOGENESIS_UP Up-regulated … STUB1 angioge…
# 5 C2:CGP Chemical and Genetic Perturbations HU_ANGIOGENESIS_UP Up-regulated … FAH angioge…
# 6 C2:CGP Chemical and Genetic Perturbations HU_ANGIOGENESIS_UP Up-regulated … COL7A1 angioge…
length(unique(msigdb.vessel$geneset))
# [1] 112
# convert gene sets associated with angiogenesis to list
# required by irGSEA package
msigdb.vessel <- msigdb.vessel %>%
dplyr::select(c("geneset", "symbol"))
msigdb.vessel$geneset <- factor(msigdb.vessel$geneset)
msigdb.vessel <- msigdb.vessel %>%
dplyr::group_split(geneset, .keep = F) %>%
purrr::map( ~.x %>% dplyr::pull(symbol) %>% unique(.)) %>%
purrr::set_names(levels(msigdb.vessel$geneset))
#### Оценка набора генов, связанных с ангиогенезом ####
pbmc3k.final <- irGSEA.score(object = pbmc3k.final, assay = "RNA", slot = "data",
custom = T, geneset = msigdb.vessel,
method = c("AUCell","UCell","singscore","ssgsea", "JASMINE", "viper"),
kcdf = 'Gaussian')
### gmt file ###
# download gmt file
gmt_url <- "https://data.broadinstitute.org/gsea-msigdb/msigdb/release/2023.2.Hs/msigdb.v2023.2.Hs.symbols.gmt"
local_gmt <- "./msigdb.v2023.2.Hs.symbols.gmt"
download.file(gmt_url , local_gmt)
msigdb <- clusterProfiler::read.gmt("./msigdb.v2023.2.Hs.symbols.gmt")
# convert to list[hallmarker] required by irGSEA package
msigdb.h <- msigdb %>%
dplyr::filter(str_detect(term, pattern = regex("HALLMARK_", ignore_case=TRUE)))
msigdb.h$term <- factor(msigdb.h$term)
msigdb.h <- msigdb.h %>%
dplyr::group_split(term, .keep = F) %>%
purrr::map( ~.x %>% dplyr::pull(gene) %>% unique(.)) %>%
purrr::set_names(levels(msigdb.h$term))
#### Оценка набора генов Hallmark ####
pbmc3k.final <- irGSEA.score(object = pbmc3k.final, assay = "RNA", slot = "data",
custom = T, geneset = msigdb.h,
method = c("AUCell","UCell","singscore","ssgsea", "JASMINE", "viper"),
kcdf = 'Gaussian')
# convert to list[go bp] required by irGSEA package
msigdb.go.bp <- msigdb %>%
dplyr::filter(str_detect(term, pattern = regex("GOBP_", ignore_case=TRUE)))
msigdb.go.bp$term <- factor(msigdb.go.bp$term)
msigdb.go.bp <- msigdb.go.bp %>%
dplyr::group_split(term, .keep = F) %>%
purrr::map( ~.x %>% dplyr::pull(gene) %>% unique(.)) %>%
purrr::set_names(levels(msigdb.go.bp$term))
#### Оценка набора генов GO-BP ####
pbmc3k.final <- irGSEA.score(object = pbmc3k.final, assay = "RNA", slot = "data",
custom = T, geneset = msigdb.go.bp,
method = c("AUCell","UCell","singscore","ssgsea", "JASMINE", "viper"),
kcdf = 'Gaussian')
# convert to list[KEGG] required by irGSEA package
msigdb.kegg <- msigdb %>%
dplyr::filter(str_detect(term, pattern = regex("KEGG_", ignore_case=TRUE)))
msigdb.kegg$term <- factor(msigdb.kegg$term)
msigdb.kegg <- msigdb.kegg %>%
dplyr::group_split(term, .keep = F) %>%
purrr::map( ~.x %>% dplyr::pull(gene) %>% unique(.)) %>%
purrr::set_names(levels(msigdb.kegg$term))
#### Оценка набора генов KEGG ####
pbmc3k.final <- irGSEA.score(object = pbmc3k.final, assay = "RNA", slot = "data",
custom = T, geneset = msigdb.kegg,
method = c("AUCell","UCell","singscore","ssgsea", "JASMINE", "viper"),
kcdf = 'Gaussian')
③irGSEA использует пакет ClusterProfiler для оценки того же набора генов.
#### work with clusterProfiler package ####
# load library
library(clusterProfiler)
library(tidyverse)
### kegg ###
# download kegg pathway (human) and write as gson file
kk <- clusterProfiler::gson_KEGG(species = "hsa")
gson::write.gson(kk, file = "./KEGG_20231128.gson")
# read gson file
kk2 <- gson::read.gson("./KEGG_20231123.gson")
# Convert to a data frame
kegg.list <- dplyr::left_join(kk2@gsid2name,
kk2@gsid2gene,
by = "gsid")
head(kegg.list)
# gsid name gene
# 1 hsa01100 Metabolic pathways 10
# 2 hsa01100 Metabolic pathways 100
# 3 hsa01100 Metabolic pathways 10005
# 4 hsa01100 Metabolic pathways 10007
# 5 hsa01100 Metabolic pathways 100137049
# 6 hsa01100 Metabolic pathways 10020
# Convert gene ID to gene symbol
gene_name <- clusterProfiler::bitr(kegg.list$gene,
fromType = "ENTREZID",
toType = "SYMBOL",
OrgDb = "org.Hs.eg.db")
kegg.list <- dplyr::full_join(kegg.list,
gene_name,
by = c("gene"="ENTREZID"))
# remove NA value if exist
kegg.list <- kegg.list[complete.cases(kegg.list[, c("gene", "SYMBOL")]), ]
head(kegg.list)
# gsid name gene SYMBOL
# 1 hsa01100 Metabolic pathways 10 NAT2
# 2 hsa01100 Metabolic pathways 100 ADA
# 3 hsa01100 Metabolic pathways 10005 ACOT8
# 4 hsa01100 Metabolic pathways 10007 GNPDA1
# 5 hsa01100 Metabolic pathways 100137049 PLA2G4B
# 6 hsa01100 Metabolic pathways 10020 GNE
# convert to list required by irGSEA package
kegg.list$name <- factor(kegg.list$name)
kegg.list <- kegg.list %>%
dplyr::group_split(name, .keep = F) %>%
purrr::map( ~.x %>% dplyr::pull(SYMBOL) %>% unique(.)) %>%
purrr::set_names(levels(kegg.list$name))
head(kegg.list)
### Разобравшись с этим, вы можете выполнить оценку KEGG.
#### Оценка набора генов KEGG ####
pbmc3k.final <- irGSEA.score(object = pbmc3k.final, assay = "RNA", slot = "data",
custom = T, geneset = kegg.list,
method = c("AUCell","UCell","singscore","ssgsea", "JASMINE", "viper"),
kcdf = 'Gaussian')
### go bp ###
# download go bp (human) and write as gson file
go <- clusterProfiler::gson_GO(OrgDb = "org.Hs.eg.db", ont = "BP")
gson::write.gson(go, file = "./go_20231128.gson")
# read gson file
go2 <- gson::read.gson("./go_20231128.gson")
# Convert to a data frame
go.list <- dplyr::left_join(go2@gsid2name,
go2@gsid2gene,
by = "gsid")
head(go.list)
# gsid name gene
# 1 GO:0000001 mitochondrion inheritance <NA>
# 2 GO:0000002 mitochondrial genome maintenance 142
# 3 GO:0000002 mitochondrial genome maintenance 291
# 4 GO:0000002 mitochondrial genome maintenance 1763
# 5 GO:0000002 mitochondrial genome maintenance 1890
# 6 GO:0000002 mitochondrial genome maintenance 2021
# Convert gene ID to gene symbol
go.list <- dplyr::full_join(go.list,
go2@gene2name,
by = c("gene"="ENTREZID"))
# remove NA value if exist
go.list <- go.list[complete.cases(go.list[, c("gene", "SYMBOL")]), ]
head(go.list)
# gsid name gene SYMBOL
# 2 GO:0000002 mitochondrial genome maintenance 142 PARP1
# 3 GO:0000002 mitochondrial genome maintenance 291 SLC25A4
# 4 GO:0000002 mitochondrial genome maintenance 1763 DNA2
# 5 GO:0000002 mitochondrial genome maintenance 1890 TYMP
# 6 GO:0000002 mitochondrial genome maintenance 2021 ENDOG
# 7 GO:0000002 mitochondrial genome maintenance 3980 LIG3
# convert to list required by irGSEA package
go.list$name <- factor(go.list$name)
go.list <- go.list %>%
dplyr::group_split(name, .keep = F) %>%
purrr::map( ~.x %>% dplyr::pull(SYMBOL) %>% unique(.)) %>%
purrr::set_names(levels(go.list$name))
head(go.list)
#### Оценка набора генов GO-BP ####
pbmc3k.final <- irGSEA.score(object = pbmc3k.final, assay = "RNA", slot = "data",
custom = T, geneset = go.list,
method = c("AUCell","UCell","singscore","ssgsea", "JASMINE", "viper"),
kcdf = 'Gaussian')
4. Завершение процесса irGSEA
library(Seurat)
library(SeuratData)
library(RcppML)
library(irGSEA)
data("pbmc3k.final")
# Оценка генного набора
pbmc3k.final <- irGSEA.score(object = pbmc3k.final,assay = "RNA",
slot = "data", seeds = 123, ncores = 1,
min.cells = 3, min.feature = 0,
custom = F, geneset = NULL, msigdb = T,
species = "Homo sapiens", category = "H",
subcategory = NULL, geneid = "symbol",
method = c("AUCell","UCell","singscore","ssgsea", "JASMINE", "viper"),
aucell.MaxRank = NULL, ucell.MaxRank = NULL,
kcdf = 'Gaussian')
# Рассчитать дифференциальные наборы генов и выполнить RRA
# Если сообщается об ошибке, рассмотрите возможность добавления параметров кода (future.globals.maxSize = 100000 * 1024^5)
result.dge <- irGSEA.integrate(object = pbmc3k.final,
group.by = "seurat_annotations",
method = c("AUCell","UCell","singscore","ssgsea", "JASMINE", "viper"))
# Просмотр идентификации RRA обычно распознается из дифференциального набора генов в нескольких методах оценки.
geneset.show <- result.dge$RRA %>%
dplyr::filter(pvalue <= 0.05) %>%
dplyr::pull(Name) %>% unique(.)
Визуальный дисплей
1) Глобальный дисплей
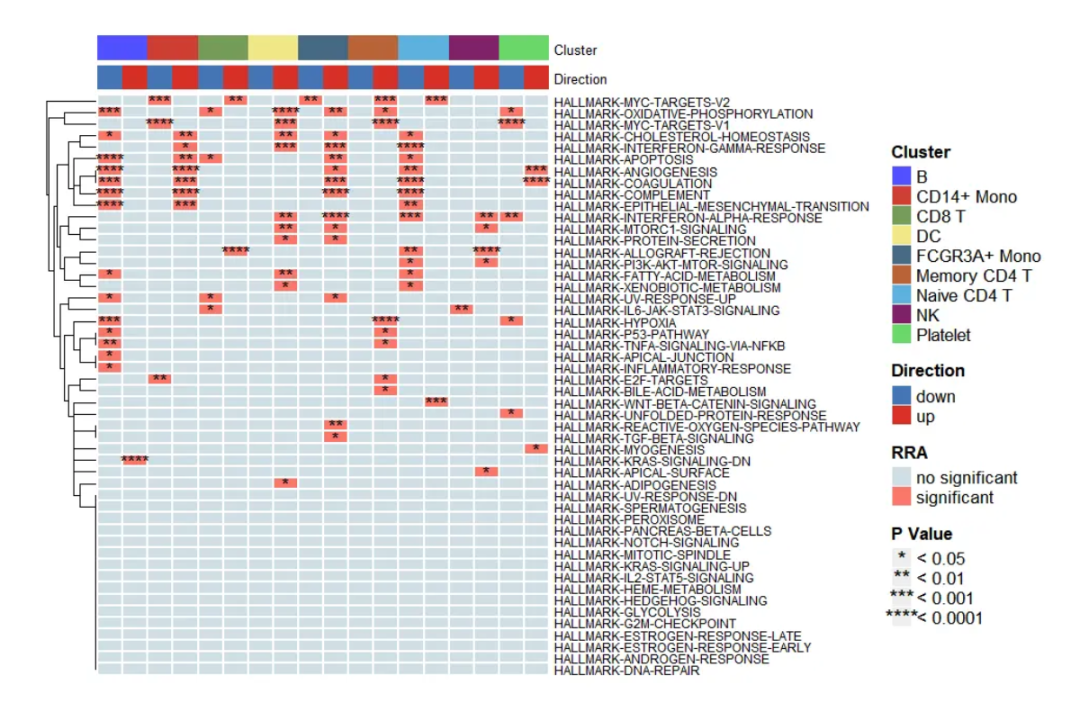
①Тепловая карта
Вы также можете изменить метод с «RRA» на «ssgsea», чтобы отобразить наборы генов с дифференциальной регуляцией или дифференциальной регуляцией с помощью конкретного метода анализа обогащения набора генов;
irGSEA.heatmap.plot <- irGSEA.heatmap(object = result.dge,
method = "RRA",
top = 50,
show.geneset = NULL)
irGSEA.heatmap.plot
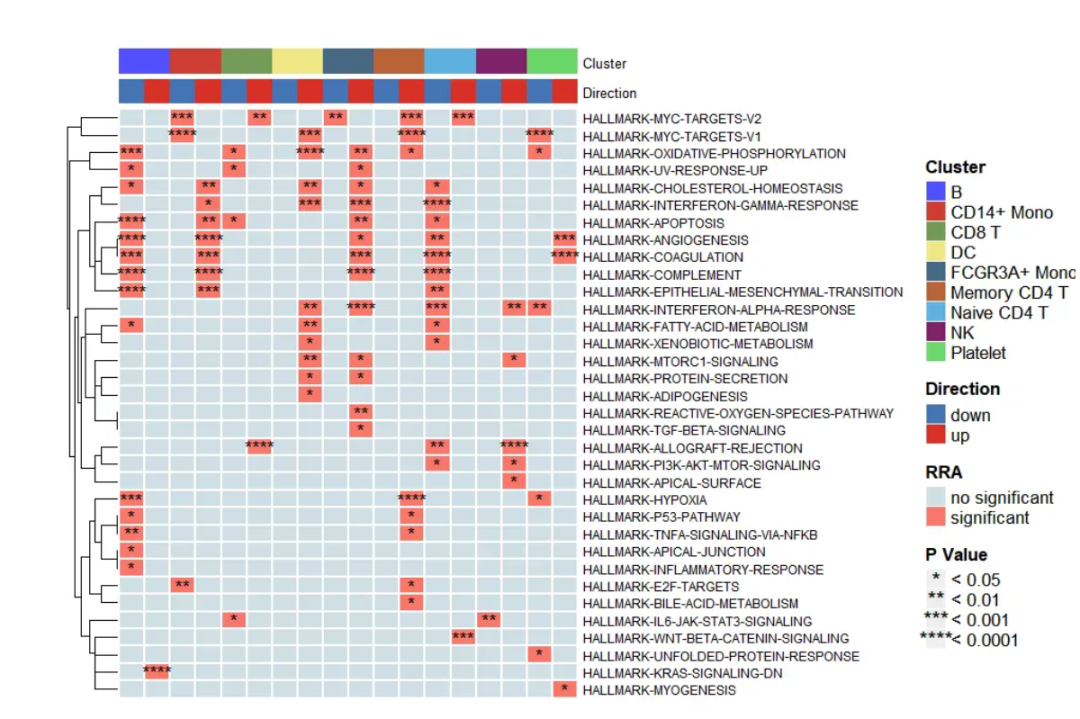
По умолчанию отображаются 50 лучших, но вы также можете отобразить те наборы генов, которые хотите отобразить. Например, я хочу показать дифференциальные наборы генов, идентифицированные RRA.
irGSEA.heatmap.plot1 <- irGSEA.heatmap(object = result.dge,
method = "RRA",
show.geneset = geneset.show)
irGSEA.heatmap.plot1
②Пузырьковая диаграмма
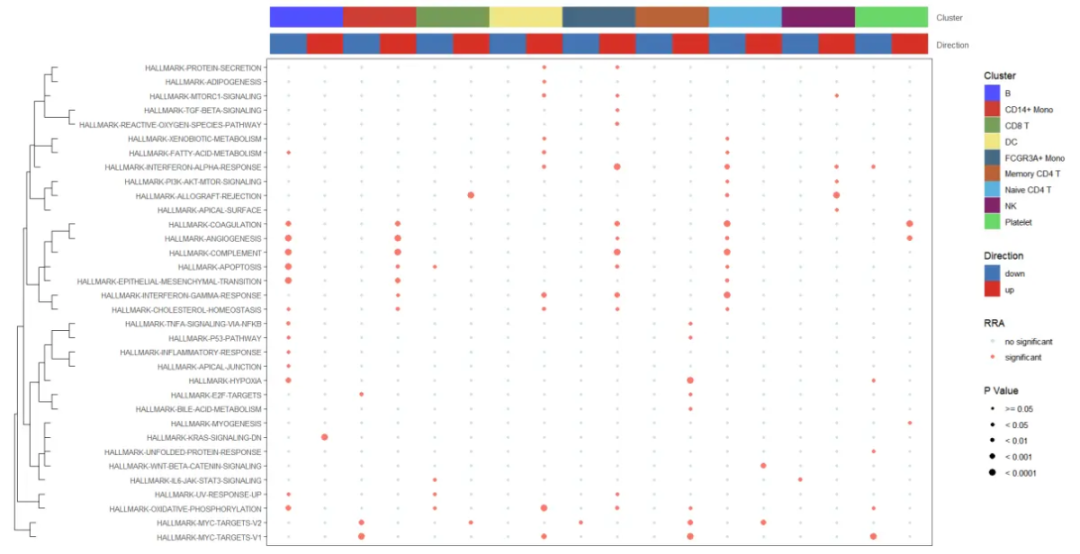
irGSEA.bubble.plot <- irGSEA.bubble(object = result.dge,
method = "RRA",
show.geneset = geneset.show)
irGSEA.bubble.plot
③upset plot
График нарушений показывает количество наборов генов со статистически значимыми различиями в каждой субпопуляции клеток при комплексной оценке, а также количество наборов дифференциальных генов с пересечением между различными субпопуляциями клеток;
irGSEA.upset.plot <- irGSEA.upset(object = result.dge,
method = "RRA",
mode = "intersect",
upset.width = 20,
upset.height = 10,
set.degree = 2,
pt_size = grid::unit(2, "mm"))
irGSEA.upset.plot
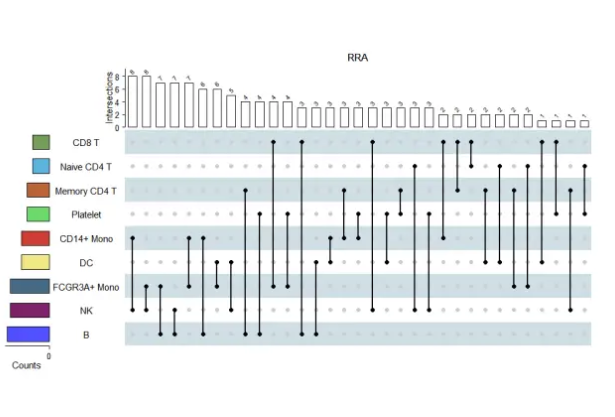
Гистограммы разных цветов слева представляют различные субпопуляции клеток; гистограмма выше представляет количество дифференциальных наборов генов с пересечением. Одна точка на пузырьковой диаграмме в середине представляет одну субпопуляцию клеток, а несколько точек, соединенных линиями; представляют несколько ячеек. Пересечение подгрупп () показывает только попарное пересечение;
④Сложенная гистограмма
Составная гистограмма конкретно показывает количество наборов генов с повышенной, пониженной регуляцией и отсутствием статистически значимых наборов генов в каждой субпопуляции клеток для каждого метода анализа обогащения набора генов;
irGSEA.barplot.plot <- irGSEA.barplot(object = result.dge,
method = c("AUCell", "UCell", "singscore",
"ssgsea", "JASMINE", "viper", "RRA"))
irGSEA.barplot.plot
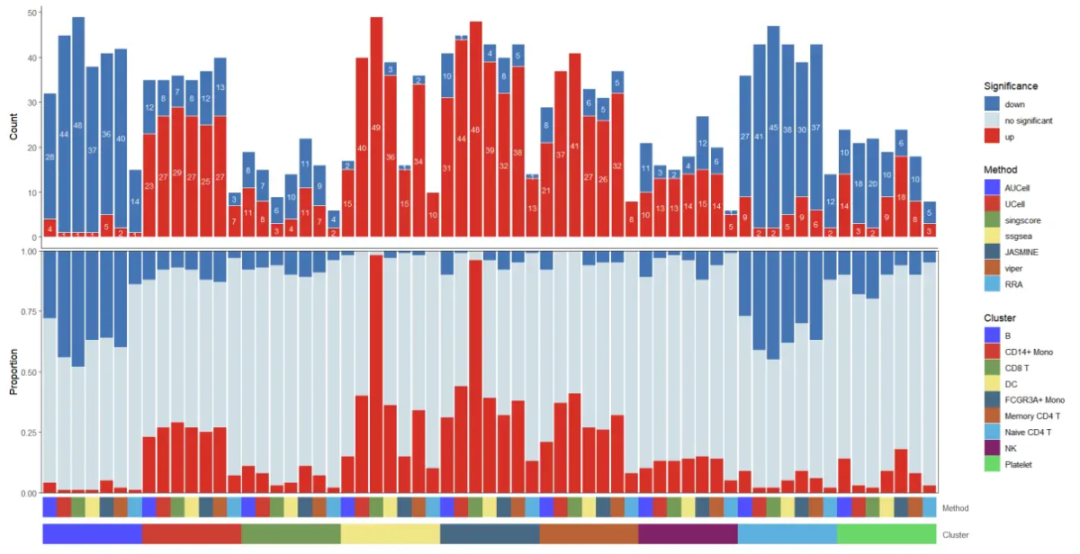
Верхняя полоса представляет количество дифференциальных генов в каждой субпопуляции, причем красный цвет представляет набор дифференциальных генов с повышенным уровнем экспрессии, а синий представляет набор дифференциальных генов с пониженной регуляцией, средний столбец представляет набор дифференциальных генов с повышенным уровнем экспрессии в каждой субпопуляции. доля наборов генов, уровень регуляции которых снижен и не имеет статистической значимости;
2) Частичное отображение
①График рассеяния плотности
Диаграммы рассеяния плотности объединяют показатели обогащения наборов генов и проекцию субпопуляций клеток в низкомерное пространство, чтобы продемонстрировать уровни пространственной экспрессии конкретных наборов генов.
scatterplot <- irGSEA.density.scatterplot(object = pbmc3k.final,
method = "UCell",
show.geneset = "HALLMARK-INFLAMMATORY-RESPONSE",
reduction = "umap")
scatterplot

Среди них, чем желтее цвет, тем выше показатель плотности, а это означает, что тем выше показатель обогащения;
②Схема полускрипки
Сюжет полускрипки показан как в виде скрипичного сюжета (слева), так и в виде коробчатого сюжета (справа). Разные цвета представляют разные субпопуляции клеток;
halfvlnplot <- irGSEA.halfvlnplot(object = pbmc3k.final,
method = "UCell",
show.geneset = "HALLMARK-INFLAMMATORY-RESPONSE")
halfvlnplot
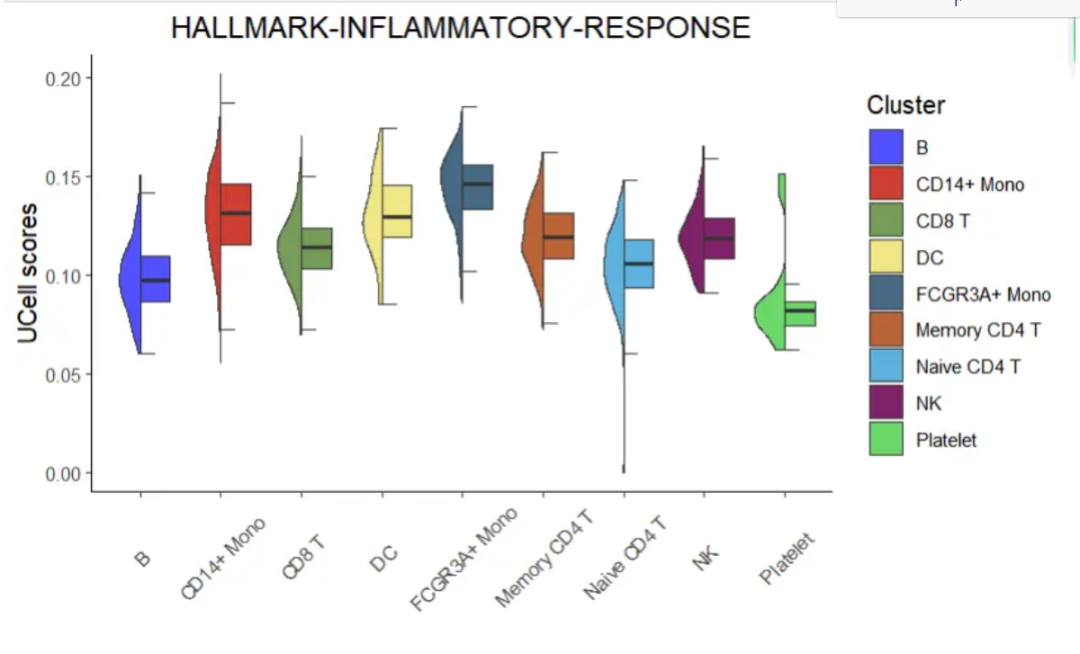
Кроме этого, это нормально
vlnplot <- irGSEA.vlnplot(object = pbmc3k.final,
method = c("AUCell", "UCell", "singscore", "ssgsea",
"JASMINE", "viper"),
show.geneset = "HALLMARK-INFLAMMATORY-RESPONSE")
vlnplot
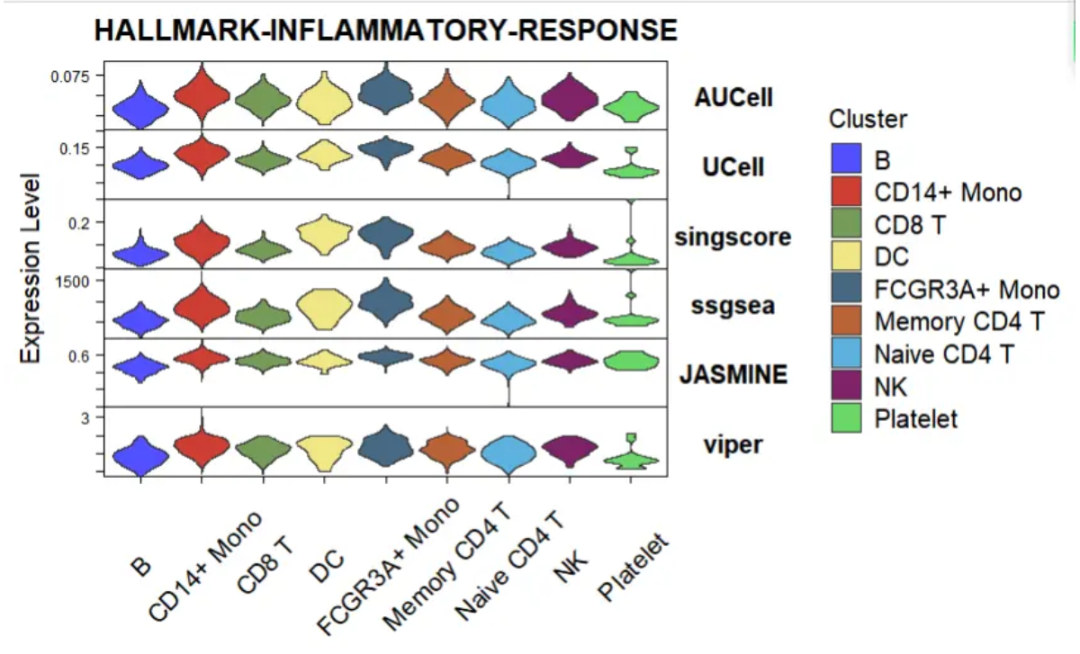
③Карта гор
Верхняя кривая плотности ядра на горном графике показывает основное распределение данных, а нижний график штрих-кода показывает конкретное количество клеточных субпопуляций. Разные цвета представляют разные субпопуляции клеток, а абсцисса представляет разные уровни экспрессии;
ridgeplot <- irGSEA.ridgeplot(object = pbmc3k.final,
method = "UCell",
show.geneset = "HALLMARK-INFLAMMATORY-RESPONSE")
ridgeplot
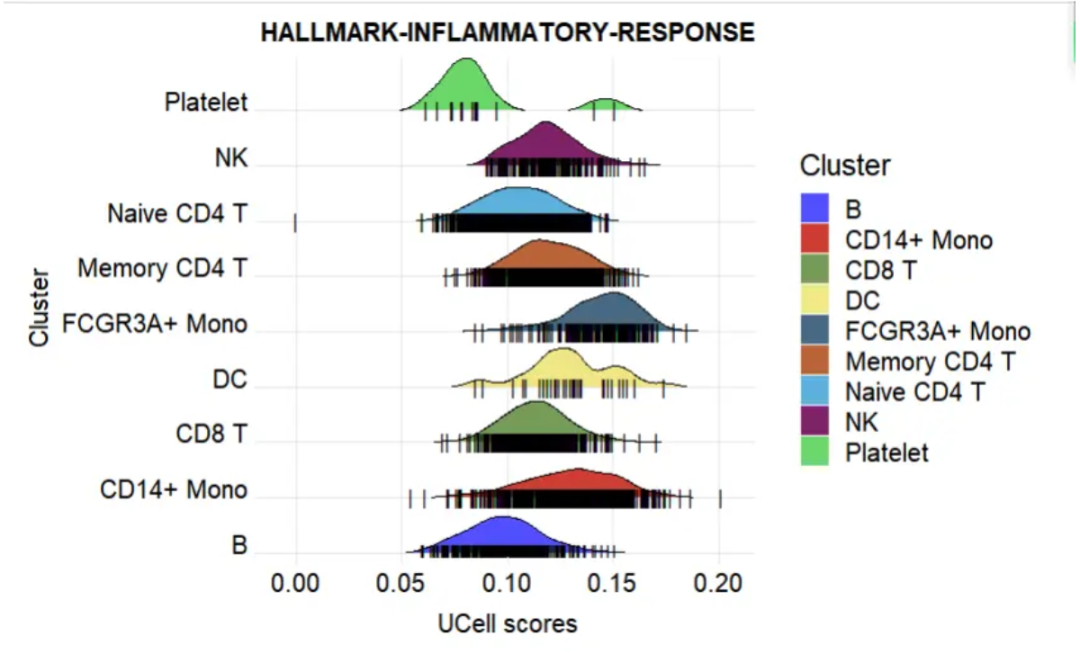
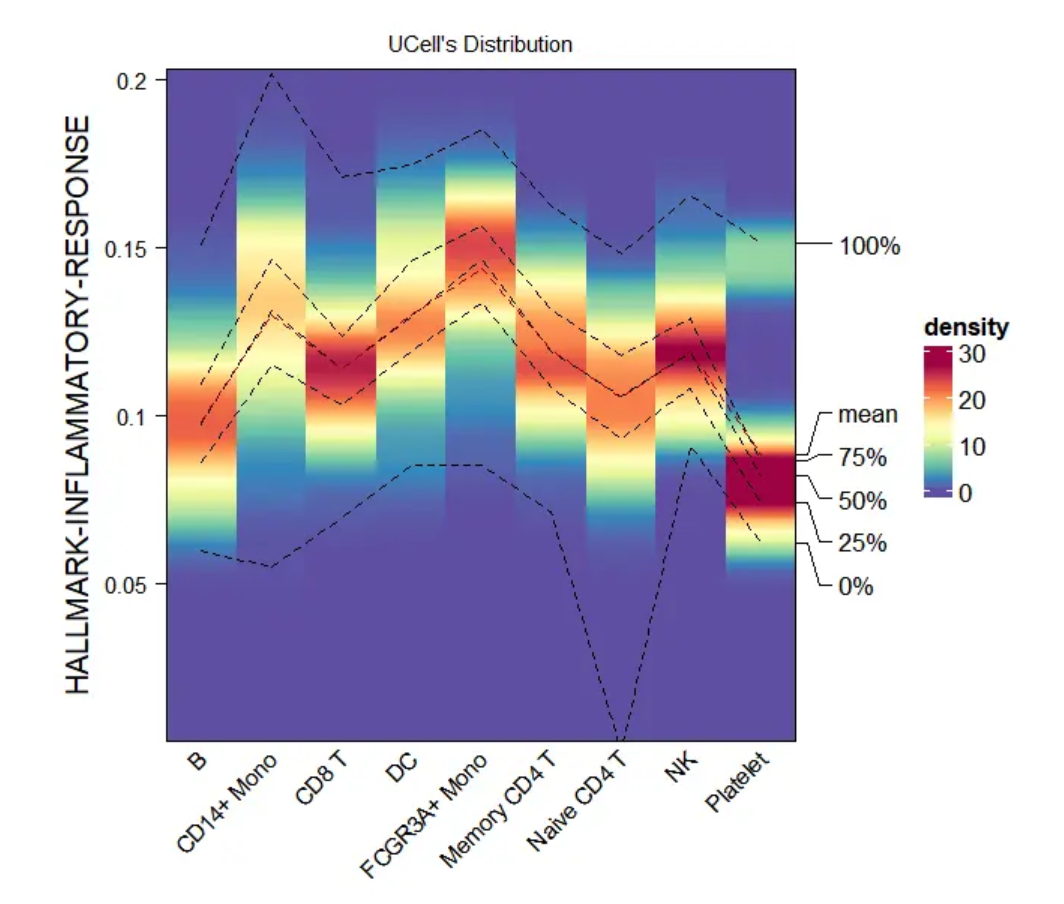
④Тепловая карта плотности
Тепловая карта плотности показывает уровни экспрессии и распределения конкретных дифференциальных генов в различных субпопуляциях клеток. Чем краснее цвет, тем выше показатель обогащения;
densityheatmap <- irGSEA.densityheatmap(object = pbmc3k.final,
method = "UCell",
show.geneset = "HALLMARK-INFLAMMATORY-RESPONSE")
densityheatmap
Ссылки
[1]
AUCell: https://doi.org/10.1038/nmeth.4463
[2]
UCell: https://doi.org/10.1016/j.csbj.2021.06.043
[3]
singscore: https://doi.org/10.1093/nar/gkaa802
[4]
ssgsea: https://doi.org/10.1038/nature08460
[5]
JASMINE: https://doi.org/10.7554/eLife.71994
[6]
VAM: https://doi.org/10.1093/nar/gkaa582
[7]
scSE: https://doi.org/10.1093/nar/gkz601
[8]
VISION: https://doi.org/10.1038/s41467-019-12235-0
[9]
wsum: https://doi.org/10.1093/bioadv/vbac016
[10]
wmean: https://doi.org/10.1093/bioadv/vbac016
[11]
mdt: https://doi.org/10.1093/bioadv/vbac016
[12]
viper: https://doi.org/10.1038/ng.3593
[13]
GSVApy: https://doi.org/10.1038/ng.3593
[14]
gficf: https://doi.org/10.1093/nargab/lqad024
[15]
GSVA: https://doi.org/10.1186/1471-2105-14-7
[16]
zscore: https://doi.org/10.1371/journal.pcbi.1000217
[17]
plage: https://doi.org/10.1186/1471-2105-6-225
[18]
ssGSEApy: https://doi.org/10.1093/bioinformatics/btac757
[19]
viperpy: https://doi.org/10.1093/bioinformatics/btac757
[20]
AddModuleScore: https://doi.org/10.1126/science.aad0501
[21]
pagoda2: https://doi.org/10.1038/nbt.4038
[22]
Sargent: https://doi.org/10.1016/j.mex.2023.102196



Неразрушающее увеличение изображений одним щелчком мыши, чтобы сделать их более четкими артефактами искусственного интеллекта, включая руководства по установке и использованию.
Копикодер: этот инструмент отлично работает с Cursor, Bolt и V0! Предоставьте более качественные подсказки для разработки интерфейса (создание навигационного веб-сайта с использованием искусственного интеллекта).
Новый бесплатный RooCline превосходит Cline v3.1? ! Быстрее, умнее и лучше вилка Cline! (Независимое программирование AI, порог 0)

Разработав более 10 проектов с помощью Cursor, я собрал 10 примеров и 60 подсказок.
Я потратил 72 часа на изучение курсорных агентов, и вот неоспоримые факты, которыми я должен поделиться!

Идеальная интеграция Cursor и DeepSeek API
DeepSeek V3 снижает затраты на обучение больших моделей
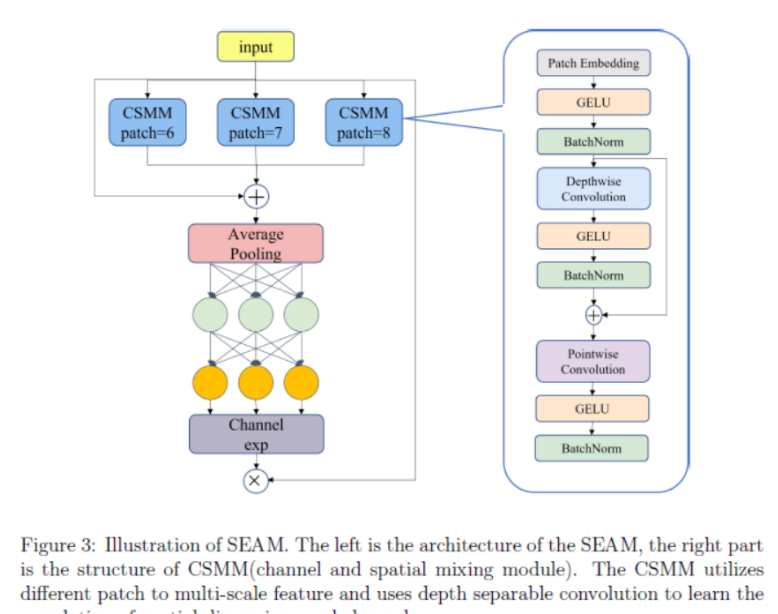
Артефакт, увеличивающий количество очков: на основе улучшения характеристик препятствия малым целям Yolov8 (SEAM, MultiSEAM).

DeepSeek V3 раскручивался уже три дня. Сегодня я попробовал самопровозглашенную модель «ChatGPT».

Open Devin — инженер-программист искусственного интеллекта с открытым исходным кодом, который меньше программирует и больше создает.
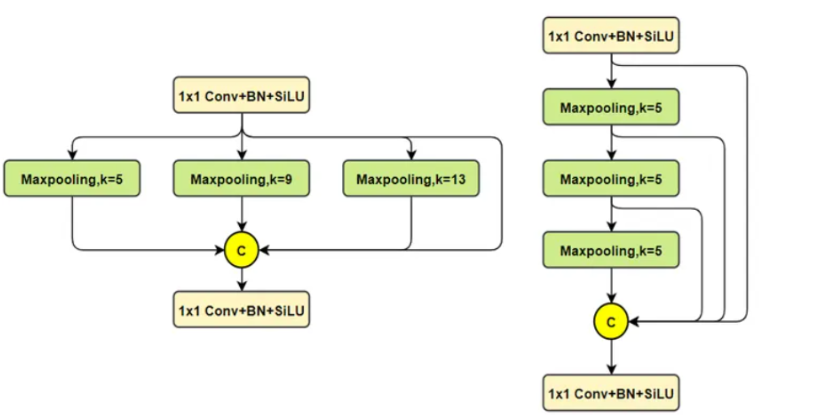
Эксклюзивное оригинальное улучшение YOLOv8: собственная разработка SPPF | SPPF сочетается с воспринимаемой большой сверткой ядра UniRepLK, а свертка с большим ядром + без расширения улучшает восприимчивое поле

Популярное и подробное объяснение DeepSeek-V3: от его появления до преимуществ и сравнения с GPT-4o.

9 основных словесных инструкций по доработке академических работ с помощью ChatGPT, эффективных и практичных, которые стоит собрать
Вызовите deepseek в vscode для реализации программирования с помощью искусственного интеллекта.
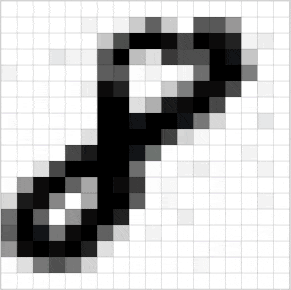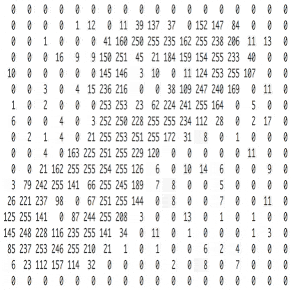
Познакомьтесь с принципами сверточных нейронных сетей (CNN) в одной статье (суперподробно)
50,3 тыс. звезд! Immich: автономное решение для резервного копирования фотографий и видео, которое экономит деньги и избавляет от беспокойства.
Cloud Native|Практика: установка Dashbaord для K8s, графика неплохая
Краткий обзор статьи — использование синтетических данных при обучении больших моделей и оптимизации производительности
MiniPerplx: новая поисковая система искусственного интеллекта с открытым исходным кодом, спонсируемая xAI и Vercel.
Конструкция сервиса Synology Drive сочетает проникновение в интрасеть и синхронизацию папок заметок Obsidian в облаке.
Центр конфигурации————Накос
Начинаем с нуля при разработке в облаке Copilot: начать разработку с минимальным использованием кода стало проще

[Серия Docker] Docker создает мультиплатформенные образы: практика архитектуры Arm64

Обновление новых возможностей coze | Я использовал coze для создания апплета помощника по исправлению домашних заданий по математике

Советы по развертыванию Nginx: практическое создание статических веб-сайтов на облачных серверах
Feiniu fnos использует Docker для развертывания личного блокнота Notepad

Сверточная нейронная сеть VGG реализует классификацию изображений Cifar10 — практический опыт Pytorch
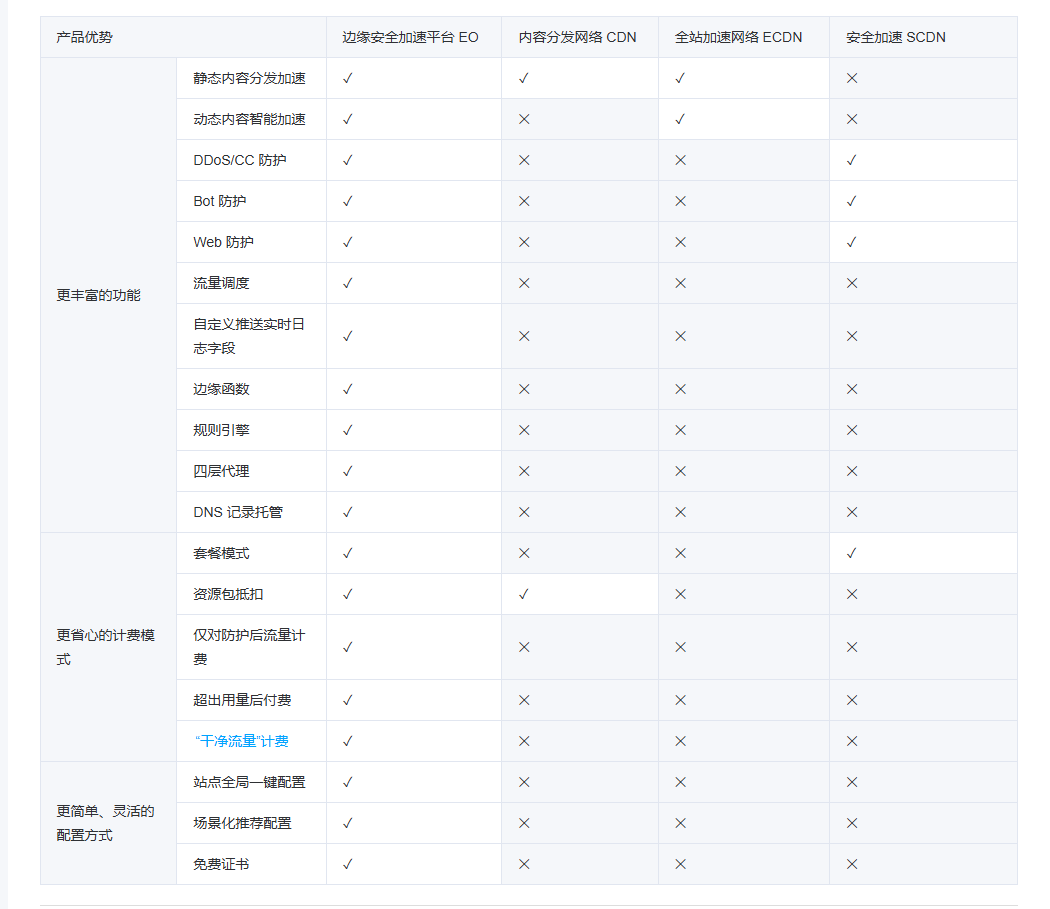
Начало работы с EdgeonePages — новым недорогим решением для хостинга веб-сайтов
[Зона легкого облачного игрового сервера] Управление игровыми архивами