Интегрированный метод анализа для нескольких наборов данных экспрессии в базе данных GEO (чип экспрессии и секвенирование транскриптома)
Во время раскопок данных,Мы часто отфильтровываем более одного набора данных, который соответствует нашим ожиданиям.,Этиданные Наборы происходят от разных исследователей。Наборы данных, полученные таким образом, будут иметь то, что мы называем пакетными эффектами, например, разное время эксперимента, разные экспериментальные партии, разные методы обработки, разные платформы секвенирования и т. д.столкнулся с такой ситуацией,Как мы выбираем данные и обращаемся с ними? Если мы выберем для анализа только один набор данных,Кажется, немного сложно объяснить выводы других исследователей.,Но если все наборы данных, соответствующие нашим экспериментальным целям, можно проанализировать, начать будет немного сложно. здесь,Давайте представим облик нескольких ГЕОданных наборов,Как нам с этим справиться?
первый,Давайте внесем ясность,Сколько наборов данных можно собрать, соответствующих нашим экспериментальным целям?,Используйте как можно больше,Потому что есть некоторые экспериментальные ошибки при анализе отдельных наборов данных.,Не репрезентативный. Во-вторых,Для нескольких эпизодов данных,У нас могут быть две идеи по проведению комплексного анализа.:Первый — объединить и удалить эти пакетные эффекты; второй — обработать каждый набор данных отдельно, а затем найти пересечение для получения общего результата.
1. Объединение и удаление пакетных эффектов
Методы объединения и удаления методов пакетной коррекции в наборах данных GEO в основном включают метод ComBat (параметрический априорный метод, ComBat_p и непараметрический метод, ComBat_n), анализ суррогатных переменных (SVA) и методы, основанные на соотношениях (метод на основе геометрического соотношения). (Ratio_G), методы центрирования среднего значения (PAMR) и дискриминации по расстоянию (DWD). По различным показателям ComBat в целом превосходит другие методы по точности, аккуратности и общей производительности. Пакет SVA в R имеет функции ComBat и ComBat_seq, которые можно использовать для коррекции пакетных эффектов. Входные данные представляют собой чистые стандартизированные данные выражения (такие как FPKM, TPM и т. д.), обычно данные чипа.
Пример кода выглядит следующим образом
# импортироватьданныенабор1
data1 <- read.csv("PRJNA423456_FPKM.csv", header = TRUE) #Набор данных здесь представляет собой смоделированный набор данных. Измените его на свой собственный набор данных для анализа.
# Импорт данных. Эпизод 2.
данные2 <- read.csv("PRJNA777728_FPKM.csv", header = TRUE) #Набор данных здесь представляет собой смоделированный набор данных. Измените его на свой собственный набор данных для анализа.
# импортироватьданныенабор3
data3 <- read.csv("PRJNA6878999_FPKM.csv", header = TRUE) #Набор данных здесь представляет собой смоделированный набор данных. Измените его на свой собственный набор данных для анализа.
# импортироватьданныенабор4
data4 <- read.csv("PRJNA590000_FPKM.csv", header = TRUE) #Набор данных здесь представляет собой смоделированный набор данных. Измените его на свой собственный набор данных для анализа.
#### слитьданныенабор
library("sva") #Нетпрограммное программное обеспечение, пожалуйста, сначала установите программное обеспечение обеспечение
library("tidyverse") #Нетпрограммное программное обеспечение, пожалуйста, сначала установите программное обеспечение обеспечение
merge_eset1 <- inner_join(data1, data2, by = "Gene.ID", suffix = c(".data1", ".data2")) #X — это имя первого столбца. Имя гена должно быть помещено в первый столбец, а не стать именем строки.
merge_eset1
merge_eset2 <- inner_join(data3, data4, by = "Gene.ID", suffix = c(".data3", ".data4"))
merge_eset <- inner_join(merge_eset1, merge_eset2, by = "Gene.ID", suffix = c(".merge_eset1", ".merge_eset2"))
merge_eset
rownames(merge_eset) <- merge_eset$Gene.ID
merge_eset <- merge_eset[,-1] #Помещаем первый столбец с именами генов в качестве имен строк
dim(merge_eset) #Viewданные измерения
exp <- as.matrix(merge_eset)
dimnames <- list(rownames(exp),colnames(exp))
data <- matrix(as.numeric(as.matrix(exp)),nrow=nrow(exp),dimnames=dimnames)
dim(data)
class(data) #Вернем «матрицу» "array"
write.csv(data,"mrna_nocombat.csv")
#Отфильтровать гены с низкой экспрессией
Expr <- data[rowSums(data)>1,]
Expr=log2(Expr+1)
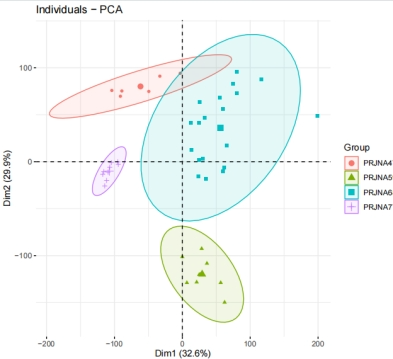
Анализ #PCA визуализирует эффект кластеризации отдельных наборов данных перед их разделением на пакеты.
# Здесь мы сначала получаем информацию о группе
group_list <- data.frame(
sample = colnames(exp_all), c(rep("PRJNAX423456_A",3),rep("PRJNA423456_B",3),rep("PRJNA777728_A",3),rep("PRJNA777728_C",3),
rep("PRJNA777728_B",3),rep("PRJNA6878999_A",10),rep("PRJNA6878999_B",9),rep("PRJNA590000_A",3),rep("PRJNA590000_C",3),
rep("PRJNA590000_B",3)))
rownames(group_list) <- group_list$sample
colnames(group_list)[2] <- "dataset"
group <- factor(group_list$dataset)
View(group_list)
##Загрузка информации об образце, включая партии
data2 <- read.csv("All_data.csv",header = T)
data2[,2] <- as.factor(data2$Type )##type представляет различные биологические процессы
data2[,3] <- as.factor(data2$Batch )##batch представляет различную информацию о пакете
row.names(data2) <- data2$Sample
View(data2)
data2
библиотека(FactoMineR)##Если она недоступна, сначала установите ее
library(factoextra)
pdf(file = "PCA_before1.pdf",width = 7,height = 6)
pre.pca <- PCA(t(exp_all),graph = FALSE)
fviz_pca_ind(pre.pca,
geom= "point",
col.ind = data2$Batch,
addEllipses = TRUE,
legend.title="Group"
)
dev.off()
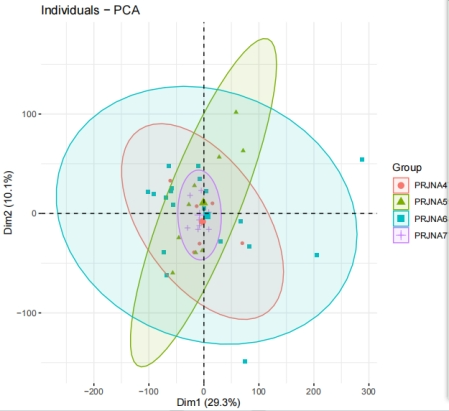
#------------------Распаковка------------------#
#### Расчет пакетных эффектов с помощью пакета sva
library(sva)
exp_all_combat <- ComBat(exp_all, batch = group_list$dataset) # партия — это информация о партии
# Ознакомьтесь с результатами удаления пакетных эффектов
library(tinyarray)
draw_pca(exp = exp_all_combat, group_list = group)
#Проведите калиброванный анализ PCA
pdf(file = "9_PCA_after.pdf",width = 7,height = 6)
pre.pca <- PCA(t(exp_all_combat),graph = FALSE)
fviz_pca_ind(pre.pca,
geom= "point",
col.ind = data2$Batch,
addEllipses = TRUE,
legend.title="Group")
dev.off()
Стоит отметить, что после удаления пакетного эффекта уровни экспрессии отдельных генов будут отрицательными. В настоящее время невозможно использовать наши традиционные EdgeR и DEseq2 для дифференциального анализа. Более того, мы использовали значение FPKM для устранения пакетного эффекта, поэтому при проведении дифференциального анализа нельзя использовать EdgeR и DEseq2, но для дифференциального анализа можно использовать пакет limma. Для анализа WGCNA это, похоже, не затронуто.
2. Интегрируйте данные и анализ
В процессе интеллектуального анализа данных мы будем анализировать данные профиля экспрессии нескольких наборов данных одновременно, чтобы получить несколько списков дифференциального анализа. Итак, как мы можем выбрать некоторые более важные или биологически значимые гены для последующих экспериментов? Традиционный подход заключается в перекрытии дифференциальных списков генов трех наборов данных, но этот метод учитывает только количество встречаемости генов и не учитывает важность генов в ранжировании нескольких дифференциальных списков.
Метод надежной агрегации рангов (RRA) может пересекать несколько ранжированных наборов генов, одновременно учитывая их ранжирование. Вообще говоря, если выбрать те гены, которые показывают различия в нескольких наборах данных, и те гены, которые занимают высокие места каждый раз, когда различия различны, их окончательный комплексный рейтинг также будет относительно высоким.
Пример кода выглядит следующим образом
setwd("F:\\RRA Analysis-2024-03-29\\4_RRA Analysis-2\\Anagen_Catagen")
#1. Считайте набор данных GSE.
files=c("PRJNA590000_A_C.DESeq2.csv",
"PRJNA779755_A_C.DESeq2.csv")
upList=list()
downList=list()
allFCList=list()
for(i in 1:length(files)){
inputFile=files[i]
rt=read.csv(inputFile)
header=unlist(strsplit(inputFile,"_"))
downList[[header[1]]]=as.vector(rt[,1])
upList[[header[1]]]=rev(as.vector(rt[,1]))
fcCol=rt[,1:2] colnames(fcCol)=c("Gene",header[[1]])
allFCList[[header[1]]]=fcCol
}
View(allFCList)
#2. Объедините все значения FC.
mergeLe=function(x,y){
merge(x,y,by="Gene",all=FALSE)}
newTab=Reduce(mergeLe,allFCList)
View(newTab)
rownames(newTab)=newTab[,1]
newTab=newTab[,2:ncol(newTab)]
newTab[is.na(newTab)]=0
dim(newTab)
#3. Получить регулируемые гены.
library(RobustRankAggreg)
upMatrix = rankMatrix(upList)
View(upMatrix)
dim(upMatrix)
upAR = aggregateRanks(rmat=upMatrix)
View(upAR)
dim(upAR)
colnames(upAR)=c("Name","Pvalue")
upAdj=p.adjust(upAR$Pvalue,method="bonferroni")
#help(p.adjust)
upXls=cbind(upAR,adjPvalue=upAdj)
upFC=newTab[as.vector(upXls[,1]),]
upXls=cbind(upXls,logFC=rowMeans(upFC))
write.table(upXls,file="up.xls",sep="\t",quote=F,row.names=F)
upSig=upXls[(upXls$Pvalue<0.01 & upXls$logFC> log2(2)),]
dim(upSig)
write.table(upSig,file="upSig_2.xls",sep="\t",quote=F,row.names=F)
#3. Получить гены с пониженной регуляцией.
downMatrix = rankMatrix(downList)
downAR = aggregateRanks(rmat=downMatrix)
colnames(downAR)=c("Name","Pvalue")
downAdj=p.adjust(downAR$Pvalue,method="BH")
downXls=cbind(downAR,adjPvalue=downAdj)
downFC=newTab[as.vector(downXls[,1]),]
downXls=cbind(downXls,logFC=rowMeans(downFC))
write.table(downXls,file="down.xls",sep="\t",quote=F,row.names=F)
downSig=downXls[(downXls$Pvalue<0.01 & downXls$logFC< -log2(2)),]
dim(downSig)
write.table(downSig,file="downSig_2.xls",sep="\t",quote=F,row.names=F)
#Интегрируйте гены с повышающей и понижающей регуляцией
allSig = rbind(upSig,downSig)
colnames(allSig)
allSig = allSig[,c("Name","logFC")]
View(allSig)
write.table(allSig,file = 'allSign_2.xls',sep = '\t',quote = F)
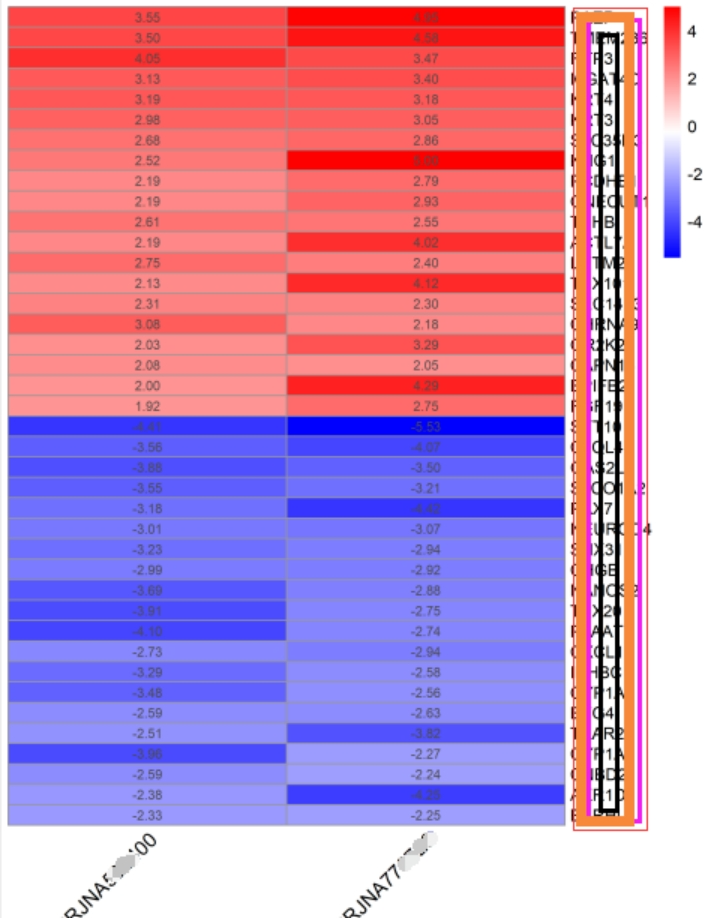
#Отображение результатов 20 лучших генов с повышающей и понижающей регуляцией logFC.tiff
hminput=newTab[c(as.vector(upSig[1:20,1]),as.vector(downSig[c(1:20),1])),]
library(pheatmap)
tiff(file="logFC2_1_2.tiff",width = 15,height = 20,units ="cm",compression="lzw",bg="white",res=400)
pheatmap(hminput,display_numbers = TRUE,fontsize_row=10,fontsize_col=12,
color = colorRampPalette(c("blue","white", "red"))(200),
cluster_cols = FALSE,cluster_rows = FALSE, angle_col = 45)
dev.off()
В настоящее время наиболее часто используемыми методами комплексного анализа являются два метода, представленные выше. Наконец, я дам вам несколько соответствующих ссылок для справки!
- Xing J, Chen M, Han Y. Multiple datasets to explore the tumor microenvironment of cutaneous squamous cell carcinoma. Math Biosci Eng. 2022 Apr 8;19(6):5905-5924. doi: 10.3934/mbe.2022276. IF: 2.6 Q2. PMID: 35603384. https://pubmed.ncbi.nlm.nih.gov/35603384/
- Li Z, Li X, Jin M, Liu Y, He Y, Jia N, Cui X, Liu Y, Hu G, Yu Q. Identification of potential biomarkers and their correlation with immune infiltration cells in schizophrenia using combinative bioinformatics strategy. Psychiatry Res. 2022 Aug;314:114658. doi: 10.1016/j.psychres.2022.114658. IF: 11.3 Q1. https://pubmed.ncbi.nlm.nih.gov/35660966/
- Mo M, Liu B, Luo Y, Tan JHJ, Zeng X, Zeng X, Huang D, Li C, Liu S, Qiu X. Construction and Comprehensive Analysis of a circRNA-miRNA-mRNA Regulatory Network to Reveal the Pathogenesis of Hepatocellular Carcinoma. Front Mol Biosci. 2022 Jan 24;9:801478. doi: 10.3389/fmolb.2022.801478. IF: 5.0 Q2. https://pubmed.ncbi.nlm.nih.gov/35141281/



Неразрушающее увеличение изображений одним щелчком мыши, чтобы сделать их более четкими артефактами искусственного интеллекта, включая руководства по установке и использованию.
Копикодер: этот инструмент отлично работает с Cursor, Bolt и V0! Предоставьте более качественные подсказки для разработки интерфейса (создание навигационного веб-сайта с использованием искусственного интеллекта).
Новый бесплатный RooCline превосходит Cline v3.1? ! Быстрее, умнее и лучше вилка Cline! (Независимое программирование AI, порог 0)

Разработав более 10 проектов с помощью Cursor, я собрал 10 примеров и 60 подсказок.
Я потратил 72 часа на изучение курсорных агентов, и вот неоспоримые факты, которыми я должен поделиться!

Идеальная интеграция Cursor и DeepSeek API
DeepSeek V3 снижает затраты на обучение больших моделей
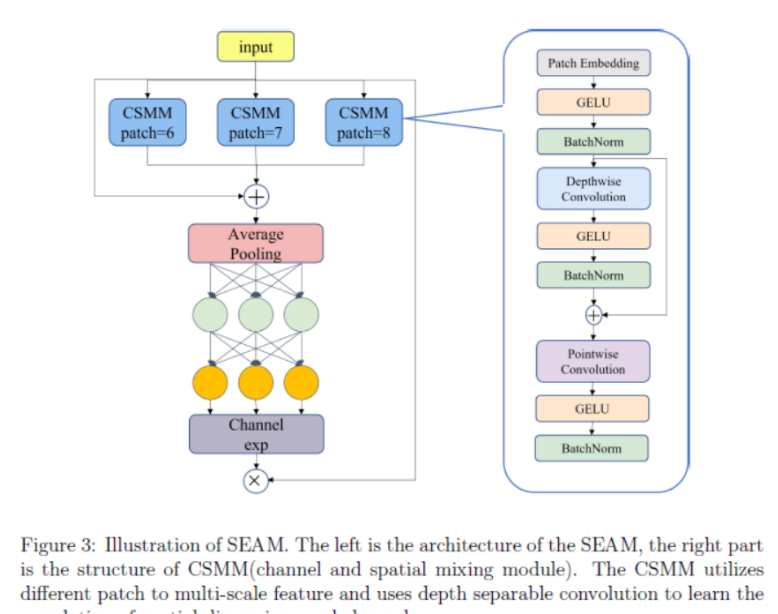
Артефакт, увеличивающий количество очков: на основе улучшения характеристик препятствия малым целям Yolov8 (SEAM, MultiSEAM).

DeepSeek V3 раскручивался уже три дня. Сегодня я попробовал самопровозглашенную модель «ChatGPT».

Open Devin — инженер-программист искусственного интеллекта с открытым исходным кодом, который меньше программирует и больше создает.
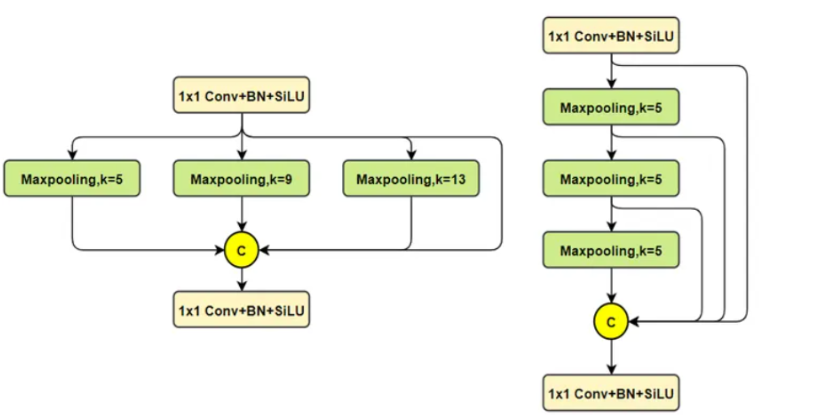
Эксклюзивное оригинальное улучшение YOLOv8: собственная разработка SPPF | SPPF сочетается с воспринимаемой большой сверткой ядра UniRepLK, а свертка с большим ядром + без расширения улучшает восприимчивое поле

Популярное и подробное объяснение DeepSeek-V3: от его появления до преимуществ и сравнения с GPT-4o.

9 основных словесных инструкций по доработке академических работ с помощью ChatGPT, эффективных и практичных, которые стоит собрать
Вызовите deepseek в vscode для реализации программирования с помощью искусственного интеллекта.
Познакомьтесь с принципами сверточных нейронных сетей (CNN) в одной статье (суперподробно)
50,3 тыс. звезд! Immich: автономное решение для резервного копирования фотографий и видео, которое экономит деньги и избавляет от беспокойства.
Cloud Native|Практика: установка Dashbaord для K8s, графика неплохая
Краткий обзор статьи — использование синтетических данных при обучении больших моделей и оптимизации производительности
MiniPerplx: новая поисковая система искусственного интеллекта с открытым исходным кодом, спонсируемая xAI и Vercel.
Конструкция сервиса Synology Drive сочетает проникновение в интрасеть и синхронизацию папок заметок Obsidian в облаке.
Центр конфигурации————Накос
Начинаем с нуля при разработке в облаке Copilot: начать разработку с минимальным использованием кода стало проще

[Серия Docker] Docker создает мультиплатформенные образы: практика архитектуры Arm64

Обновление новых возможностей coze | Я использовал coze для создания апплета помощника по исправлению домашних заданий по математике

Советы по развертыванию Nginx: практическое создание статических веб-сайтов на облачных серверах
Feiniu fnos использует Docker для развертывания личного блокнота Notepad

Сверточная нейронная сеть VGG реализует классификацию изображений Cifar10 — практический опыт Pytorch
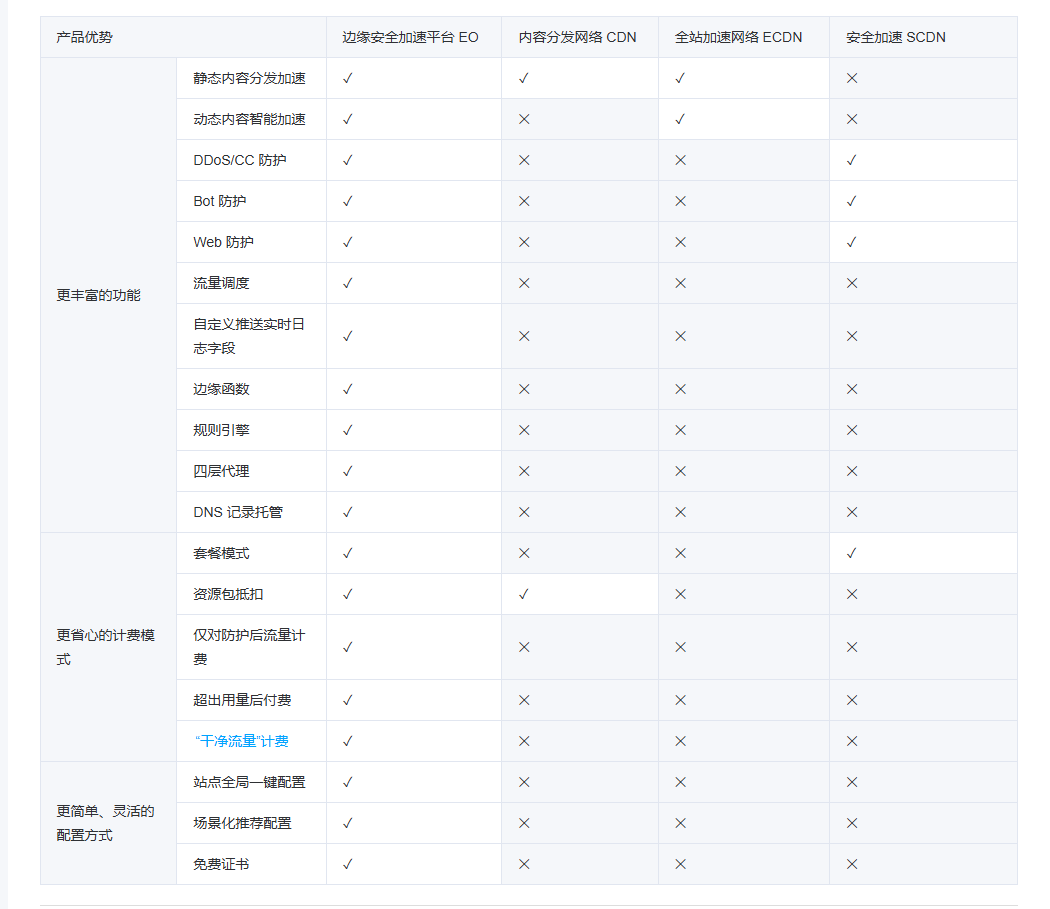
Начало работы с EdgeonePages — новым недорогим решением для хостинга веб-сайтов
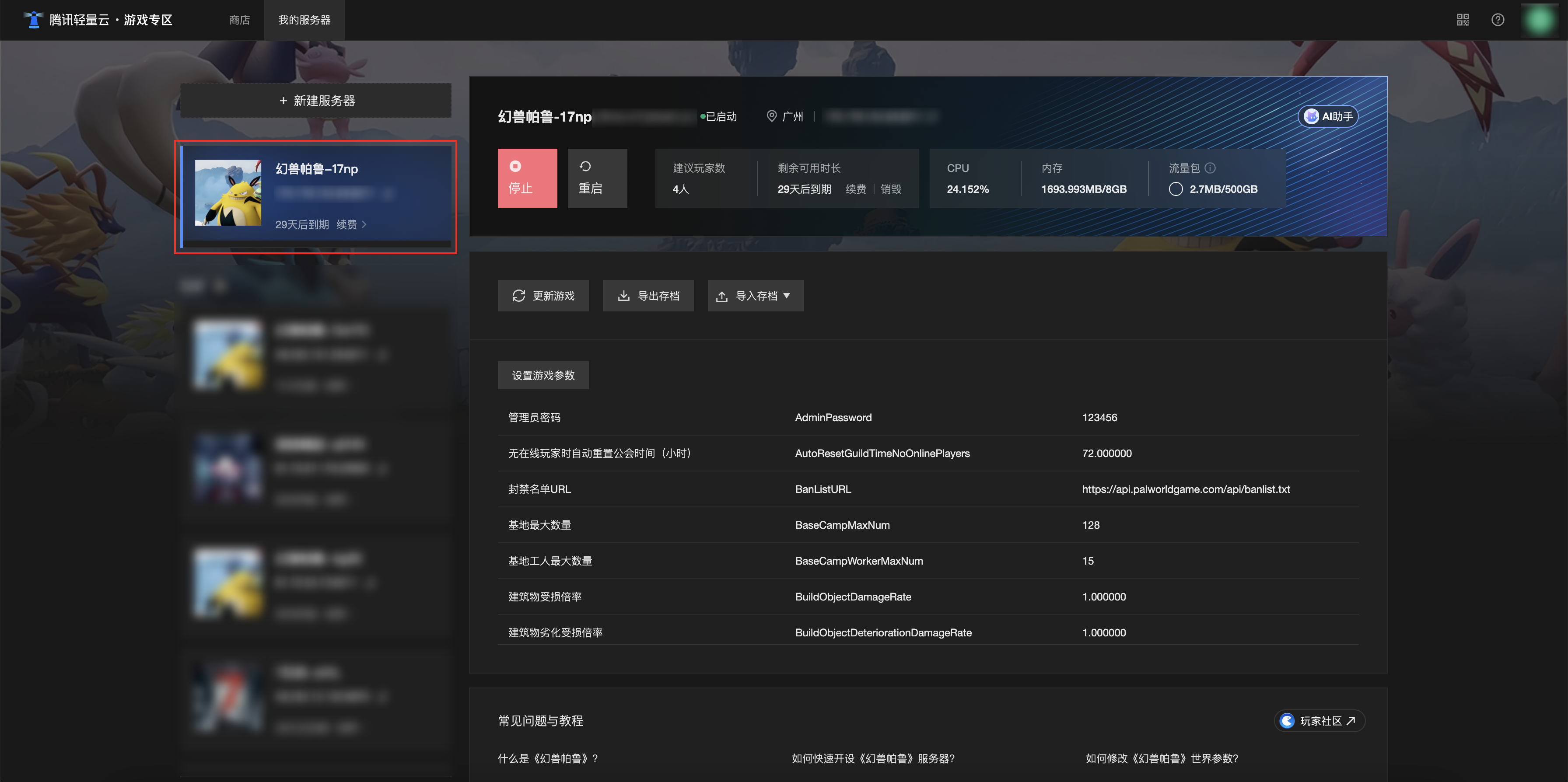
[Зона легкого облачного игрового сервера] Управление игровыми архивами