Анализ пространственных транскриптомов Сёра (1) считывание данных
Текущая одноклеточная транскриптомика в определенной степени задействована с точки зрения размера выборки, методов анализа и влажных экспериментов. Другая тенденция заключается во внедрении одноклеточной мультиомики (например, scATAC-seq и т. д.) и пространственных измерений. включая пространственную группу транскрипции, пространственный метаболом, пространственный протеом, пространственный ATAC и т. д.
Что касается изучения пространственного анализа транскриптома, я рекомендую сначала изучить анализ транскриптома одной клетки и овладеть навыками чтения данных одной клетки, рутинного анализа, интеграции и разделения пакетов, а также некоторого расширенного анализа (например, псевдохронологического анализа, фактора транскрипции). и анализ клеточной коммуникации), на этой основе можно очень быстро понять и изучить пространственный транскриптом одной клетки. Официальный документ Сёра (https://satijalab.org/seurat/articles/spatial_vignette.html) представляет собой. хороший вводный урок.
Прежде чем изучать это руководство по холостому ходу, я сначала расскажу, как считывать данные холостого хода на языке R, а затем конструировать их в объект Сёра.
1. Введение
космосданные Как хранить вSeuratсередина?
Данные Visium из 10x включают в себя следующие типы данных:
- Получите точку (пятно) через матрицу экспрессии генов
- Изображения срезов тканей(коллекцияданныечасH&Eкрашение)
- Коэффициент масштабирования исходного изображения с высоким разрешением и изображения с низким разрешением, используемого для отображения.
В объекте Сёра матрица экспрессии «Спот по генам» аналогична типичному анализу «РНК», но содержит данные на уровне пятна, а не на уровне одной клетки. Само изображение хранится в слоте изображений объекта Сёра. Слоты изображений также хранят необходимую информацию для связи пятен с их физическим расположением на изображениях тканей.
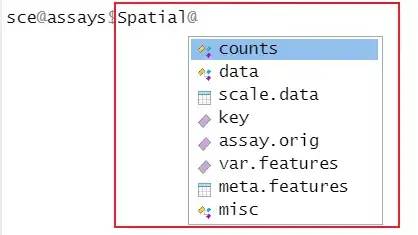
image-20230312105819088
Последующая обработка холостого хода аналогична обработке отдельных ячеек и в основном включает:
- Высадка на клеточном уровне,Считайте в R как Seuratобъект;
- Двухсотовое предсказание (необязательно);
- Низкокачественная клеточная фильтрация (опция);
- Стандартизированный выбор и нормализация функций;
- Кластеризация с уменьшением размерности;
- Групповая аннотация;
- Анализ дифференциальной экспрессии позволяет получить маркеры признаков.
Судя по аннотированным данным, последующий анализ обычно можно классифицировать как персонализированный анализ. Как и анализ отдельных клеток, он в основном включает в себя:
- Анализ обогащения: например, анализ обогащения GO, KEGG и GSEA;
- Транскрипционный факторный анализ;
- Квазихронометрический анализ холостого хода;
- Анализ простоя сотовой связи;
- ......
2. Как читать пустые данные на языке R
Шаг 1. Загрузка и установка пакета R.
library(Seurat)
library(SeuratData)
library(ggplot2)
library(patchwork)
library(dplyr)
Шаг 2. Загрузите данные.
Существуют разные стратегии загрузки для разных типов данных:
(1) Загрузите образец данных с официального сайта Seurat.
Пример данных доступен по адресу https://support.10xgenomics.com/spatial-gene-expression/datasets.,и использоватьLoad10X_Spatial()функциязагрузить его вSeurat。Это будет читатьspacerangerВыход трубы,и вернутьсяSeuratобъект,Объект содержит данные экспрессии на уровне пятна и связанные изображения срезов тканей.
Пример данных в этой статье относится к платформе 10X.,встроен вSeuratDataRСумка,Способ загрузки следующий:
InstallData("stxBrain")
brain <- LoadData("stxBrain", type = "anterior1")
brain
# An object of class Seurat
# 31053 features across 2696 samples within 1 assay
# Active assay: Spatial (31053 features, 0 variable features)
## Проверьте пример данных
head(brain@meta.data)
# orig.ident nCount_Spatial nFeature_Spatial slice region
# AAACAAGTATCTCCCA-1 anterior1 13069 4242 1 anterior
# AAACACCAATAACTGC-1 anterior1 37448 7860 1 anterior
# AAACAGAGCGACTCCT-1 anterior1 28475 6332 1 anterior
# AAACAGCTTTCAGAAG-1 anterior1 39718 7957 1 anterior
# AAACAGGGTCTATATT-1 anterior1 33392 7791 1 anterior
# AAACATGGTGAGAGGA-1 anterior1 20955 6291 1 anterior
SpatialDimPlot(brain,alpha = 0)
#Дополнительная ссылка на параметры
?SpatialDimPlot()
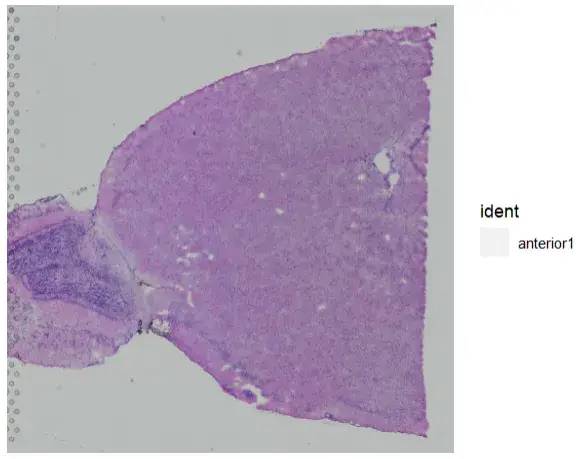
(2) Загрузите выходные данные восходящего канала 10X Cellrange.
Обычный процесс не будет использоватьсяLoadDataфункциячитатьданные,Потому что обычно мы получаем 10 X Space RangerВыходной результат:
Ниже приведены данные, загруженные с официального сайта 10X:
https://www.10xgenomics.com/resources/datasets?query=&page=1&configure%5Bfacets%5D%5B0%5D=chemistryVersionAndThroughput&configure%5Bfacets%5D%5B1%5D=pipeline.version&configure%5BhitsPerPage%5D=500&configure%5BmaxValuesPerFacet%5D=1000&menu%5Bproducts.name%5D=Spatial%20Gene%20Expression
test_data = Load10X_Spatial(data.dir = "./input/",
filename = "Visium_FFPE_Human_Normal_Prostate_filtered_feature_bc_matrix.h5",
assay = "Spatial",
slice = "test")
head(test_data@meta.data)
SpatialDimPlot(test_data,alpha = 0)
#Так же, как и данные одной ячейки, эти имена можно изменить вручную
test_data@project.name <- "test"
Idents(test_data) <- "test"
test_data$orig.ident <- "test"
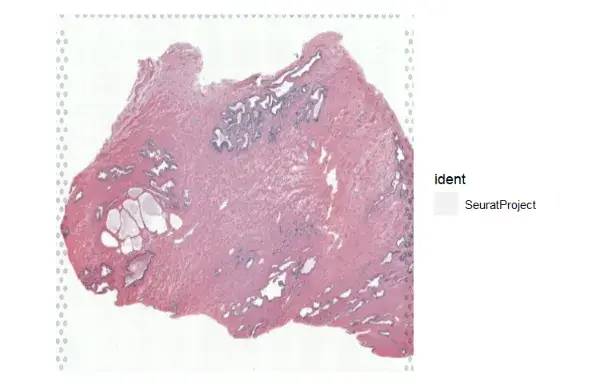
image-20230224173610241
## Получить координаты изображения среза:
img<- GetTissueCoordinates(test_data)
img$imagerow = 540-img$imagerow
write.csv(img,file = "./No_IHC/position_information.csv")
head(img)
# imagerow imagecol
# AAACAAGTATCTCCCA-1 183.3725 434.0820
# AAACAATCTACTAGCA-1 442.7995 247.5663
# AAACAGAGCGACTCCT-1 381.8799 409.0720
# AAACAGCTTTCAGAAG-1 222.4493 139.4663
# AAACCCGAACGAAATC-1 210.8508 475.3213
# AAACCGGAAATGTTAA-1 161.2021 503.7606
(3) Чтение нетрадиционных данных
3.1 Отсутствующие изображения ИГХ
Иногда данные, загруженные из базы данных, из-за отсутствия изображений ИГХ можно прочитать следующими способами:
# Считайте данные пространства как данные одной ячейки.
test_data2 = Read10X("./input/filtered_feature_bc_matrix/")
test_data2 <- CreateSeuratObject(counts = test_data2,
min.features = 0,
project = "test")
test_data2
# An object of class Seurat
# 17943 features across 2543 samples within 1 assay
Если данных IHC о холостом ходу нет, автор обычно предоставляет информацию о местоположении:
# Чтение информации о положении одной ячейки
position = read.csv("./No_IHC/position_information.csv",header = T,row.names = 1)
head(position)
position = select(position,imagecol,imagerow)
#Включить информацию о местоположении в одну ячейку seuratobject
colnames(position) = paste0("Spatial_",1:ncol(position))
test_data2[["spatial"]] <- CreateDimReducObject(embeddings = as.matrix(position),
key = "Spatial",assay = "RNA")
DimPlot(test_data2,reduction = "spatial",pt.size = 1)

3.2 При отсутствии ИГХ-изображений можно также использовать метод Slide-seq.
test_data2 = Read10X("./input/filtered_feature_bc_matrix/")
test_data2 <- CreateSeuratObject(counts = test_data2,
min.features = 0,
project = "test",
assay="Spatial")
test_data2
# An object of class Seurat
# 17943 features across 2543 samples within 1 assay
# Active assay: RNA (17943 features, 0 variable features)
coord.df = read.csv("./position_information.csv",header = T,row.names = 1)
test_data2@images$image = new(
Class = 'SlideSeq',
assay = "Spatial",
key = "image_",
coordinates = coord.df
)
test_data2
SpatialDimPlot(test_data2)
Выше приведены несколько распространенных методов чтения пустых данных на язык R.
- END -



Неразрушающее увеличение изображений одним щелчком мыши, чтобы сделать их более четкими артефактами искусственного интеллекта, включая руководства по установке и использованию.
Копикодер: этот инструмент отлично работает с Cursor, Bolt и V0! Предоставьте более качественные подсказки для разработки интерфейса (создание навигационного веб-сайта с использованием искусственного интеллекта).
Новый бесплатный RooCline превосходит Cline v3.1? ! Быстрее, умнее и лучше вилка Cline! (Независимое программирование AI, порог 0)

Разработав более 10 проектов с помощью Cursor, я собрал 10 примеров и 60 подсказок.
Я потратил 72 часа на изучение курсорных агентов, и вот неоспоримые факты, которыми я должен поделиться!

Идеальная интеграция Cursor и DeepSeek API
DeepSeek V3 снижает затраты на обучение больших моделей
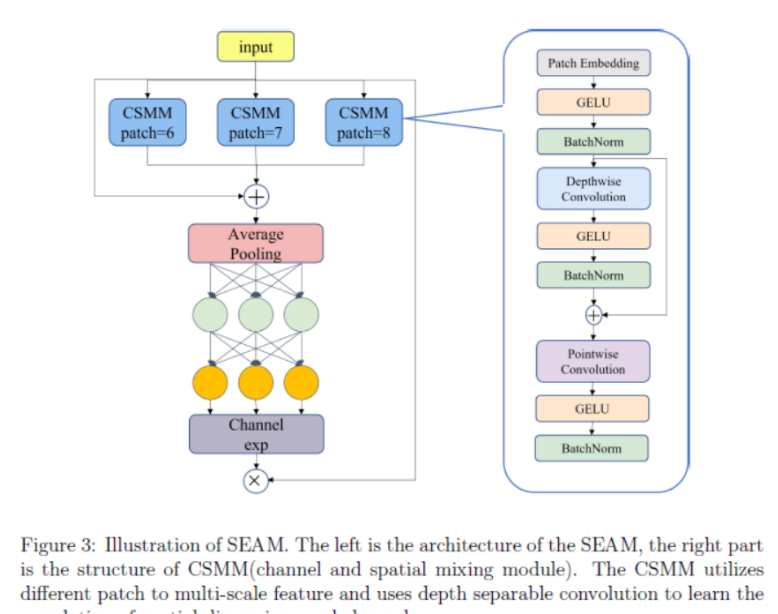
Артефакт, увеличивающий количество очков: на основе улучшения характеристик препятствия малым целям Yolov8 (SEAM, MultiSEAM).

DeepSeek V3 раскручивался уже три дня. Сегодня я попробовал самопровозглашенную модель «ChatGPT».

Open Devin — инженер-программист искусственного интеллекта с открытым исходным кодом, который меньше программирует и больше создает.
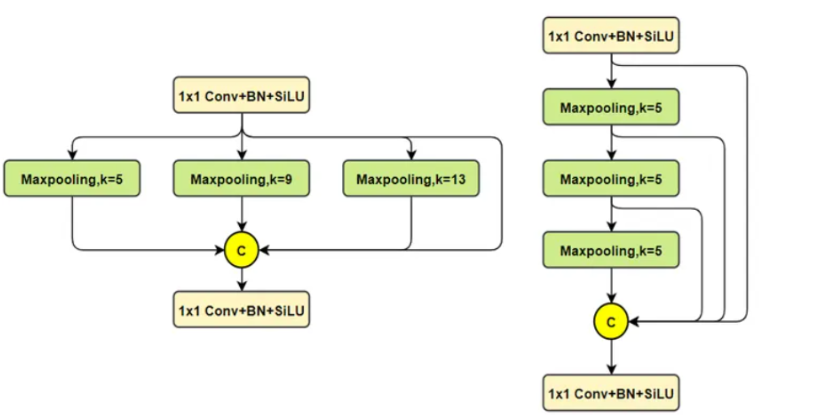
Эксклюзивное оригинальное улучшение YOLOv8: собственная разработка SPPF | SPPF сочетается с воспринимаемой большой сверткой ядра UniRepLK, а свертка с большим ядром + без расширения улучшает восприимчивое поле

Популярное и подробное объяснение DeepSeek-V3: от его появления до преимуществ и сравнения с GPT-4o.

9 основных словесных инструкций по доработке академических работ с помощью ChatGPT, эффективных и практичных, которые стоит собрать
Вызовите deepseek в vscode для реализации программирования с помощью искусственного интеллекта.
Познакомьтесь с принципами сверточных нейронных сетей (CNN) в одной статье (суперподробно)
50,3 тыс. звезд! Immich: автономное решение для резервного копирования фотографий и видео, которое экономит деньги и избавляет от беспокойства.
Cloud Native|Практика: установка Dashbaord для K8s, графика неплохая
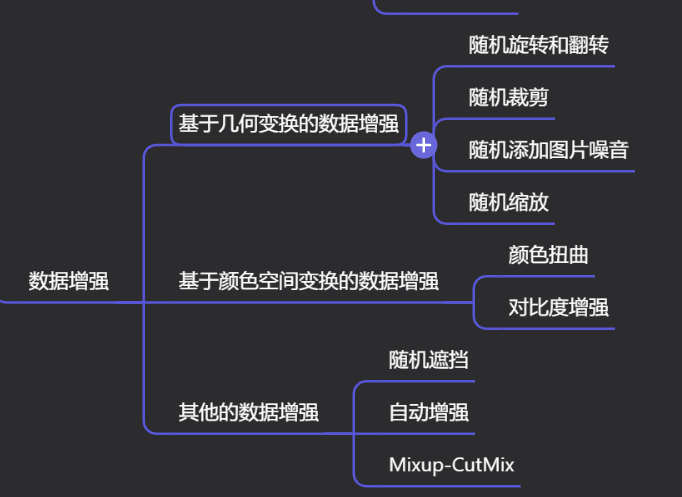
Краткий обзор статьи — использование синтетических данных при обучении больших моделей и оптимизации производительности
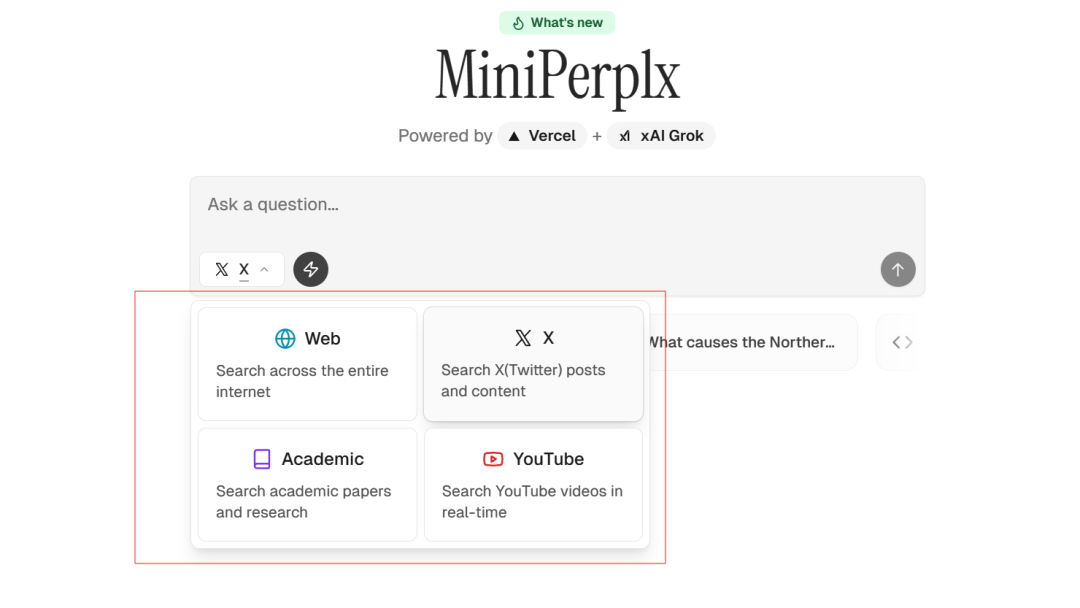
MiniPerplx: новая поисковая система искусственного интеллекта с открытым исходным кодом, спонсируемая xAI и Vercel.
Конструкция сервиса Synology Drive сочетает проникновение в интрасеть и синхронизацию папок заметок Obsidian в облаке.
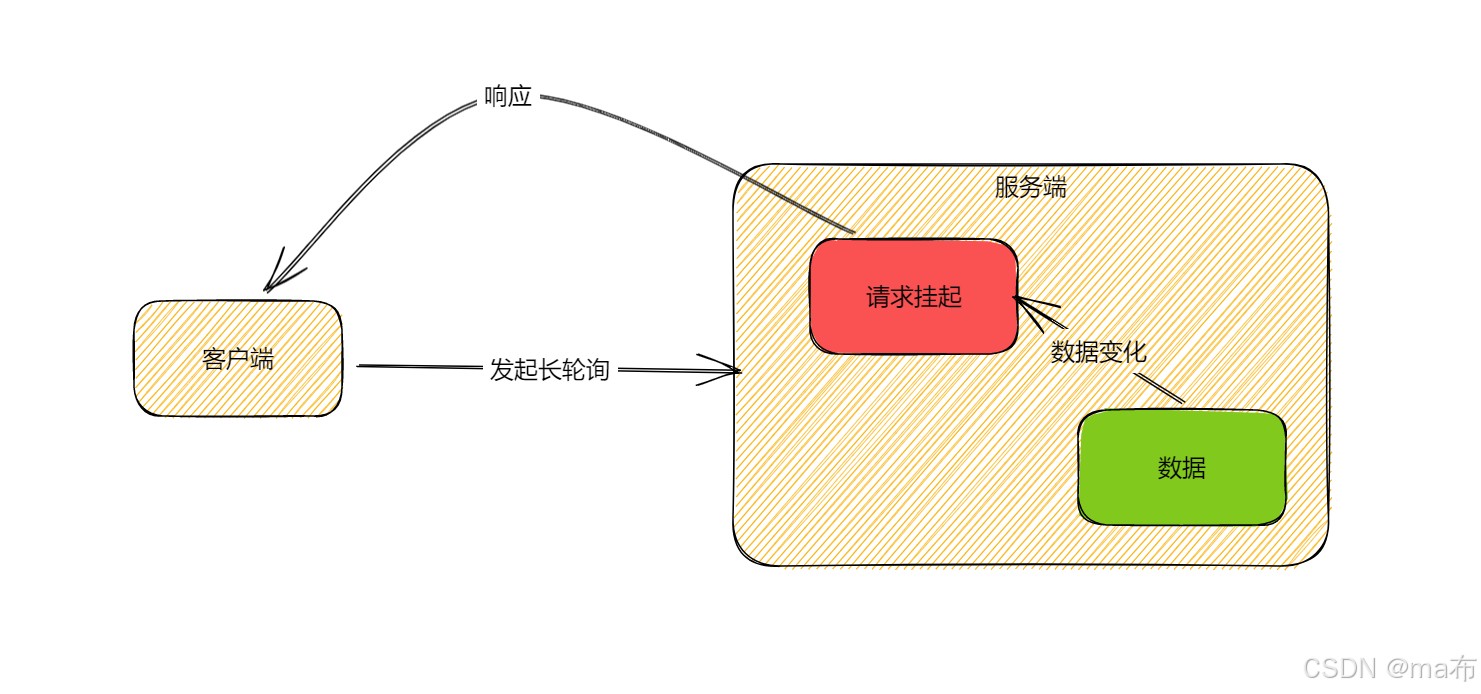
Центр конфигурации————Накос
Начинаем с нуля при разработке в облаке Copilot: начать разработку с минимальным использованием кода стало проще

[Серия Docker] Docker создает мультиплатформенные образы: практика архитектуры Arm64

Обновление новых возможностей coze | Я использовал coze для создания апплета помощника по исправлению домашних заданий по математике

Советы по развертыванию Nginx: практическое создание статических веб-сайтов на облачных серверах
Feiniu fnos использует Docker для развертывания личного блокнота Notepad

Сверточная нейронная сеть VGG реализует классификацию изображений Cifar10 — практический опыт Pytorch
Начало работы с EdgeonePages — новым недорогим решением для хостинга веб-сайтов
[Зона легкого облачного игрового сервера] Управление игровыми архивами