Анализ miloR отдельных клеток (метод дифференциального анализа численности клеток на основе графика KNN)
Обычно после того, как две или более группы образцов подвергаются разным обработкам/вмешательствам, исследователи сначала сравнивают количество клеток до и после обработки одной и той же субпопуляции клеток, но после эпохи разрешения отдельных клеток, даже если одни и те же разные клетки внутри субпопуляции также следует рассматривать разные выборки.
Тогда возникает вопрос. Поскольку их следует рассматривать как разные образцы, предполагая, что количество клеток в одной и той же субпопуляции клеток изменилось после вмешательства, является ли это изменение все еще ценным для ссылок и исследований (только с точки зрения количества клеток) ( Смотри), автор считает, что она должна представлять справочную и исследовательскую ценность. Хотя это правда, что разные клетки следует рассматривать как разные типы образцов, это различие не может скрыть присущие им свойства как большого типа клеток в биологии, и гетерогенность каждой клетки также следует учитывать. Подкатегории, основанные на характеристиках крупных клеток. категории.
Тогда некоторые люди могут запутаться. Поскольку определение клеточных субпопуляций основано на предварительных знаниях, то есть на искусственном определении клеточных субпопуляций на основе нескольких известных важных маркерных генов, то если предположить, что клеточные субпопуляции определены неправильно, произойдет ли это? вызвать проблемы в окончательных результатах дифференциального анализа? Такая ситуация должна существовать (в основном в сложных субпопуляциях клеток).
Поэтому некоторые исследователи предложили метод дифференциальной численности субпопуляций клеток, таких как miloR.,Этот метод имеет очень большое преимущество в том, что он можетИзбегайте предопределенных зависимостей кластеризации (предопределенных субпопуляций ячеек) 。
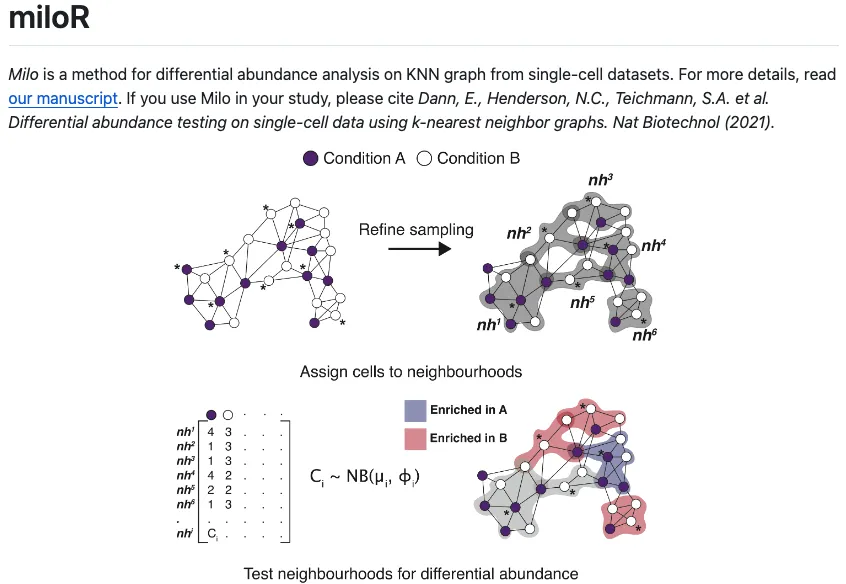
Предположим, у нас есть партия, содержащая болезнь и нормальную информацию. данные секвенирования ячейки,miloR сначала случайным образом определяет узлы ячеек (точки данных) в ячейках, а затем использует метод K-ближайших соседей (K-ближайший сосед соседи), чтобы определить взаимосвязь близости между определенным узлом и другими точками данных окружающих ячеек и найти другие точки данных (ячейки), ближайшие к этим узлам (например, метод Евклида) ,Рассматривайте эти небольшие группы ячеек со смежными отношениями как разные группы.,В этот момент мы можем представить,В каждой группе будет много клеток с болезнью и нормальной информацией.,Затем количество клеток у людей с заболеванием и без него в этих группах сравнивали и суммировали.,наконец-то окПолучайте результаты на основе внутренних тенденций ячеек в широкой категории ячеек.(Например, связана ли тенденция с направлением заболевания.)。
После получения такого результата,Независимо от того, сочетается ли это с предопределенными результатами группировки клеток или используется в качестве эталона для последующих результатов группировки клеток, это поможет пользователям лучше понять изменяющееся состояние различных субпопуляций клеток независимо от того, есть ли заболевание или нет.,Так что используйтеmiloRможно сделать эффективнодляДополнительные инструменты для клеточного дифференциального анализа на уровне отдельных клеток。
Процесс анализа
1.Импорт
rm(list = ls())
V5_path = "/Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/seurat5/"
.libPaths(V5_path)
.libPaths()
library(stringr)
library(Seurat)
library(miloR)
library(scater)
library(scran)
library(dplyr)
library(patchwork)
library(qs)
library(BiocParallel)
register(MulticoreParam(workers = 4, progressbar = TRUE))
load("scRNA.Rdata")2. Предварительная обработка данных
# Извлеките два образца
# check
table(scRNA$orig.ident)
# sample1 sample2 sample3 sample4 sample5 sample6
# 687 622 683 686 678 675
table(scRNA$celltype)
# CD4+ T-cells Fibroblasts B-cells CD8+ T-cells Neutrophils
# 1007 762 667 547 378
# Monocytes Adipocytes NK cells Endothelial cells
# 311 244 87 28
dat <- scRNA@meta.data
# Добавьте информацию о партии и преобразуйте ее в числа.
dat$batch <- ifelse(grepl("sample2|sample4|sample6", dat$orig.ident), "1", "2")
# Добавьте некоторую информацию о группировке, она создается по желанию.
dat$group <- ifelse(grepl("sample[1-3]", dat$orig.ident), "con", "treat")
scRNA@meta.data <- dat3. Создайте объект miloR.
# В связи с необходимостью ввода объекта SingleCellExperiment
# Итак, давайте сначала преобразуем его
scRNA_pre <- as.SingleCellExperiment(scRNA_pre)
# Создайте объект miloR
scRNA_pre <- Milo(scRNA_pre)
scRNA_pre
# class: Milo
# dim: 24357 1309
# metadata(0):
# assays(3): counts logcounts scaledata
# rownames(24357): AL627309.1 AL627309.3 ... AF127936.2 AF127577.3
# rowData names(0):
# colnames(1309): AAAGAACAGTCTGTAC-1_1 AAAGGGCTCTCGCGTT-1_1 ...
# TTTGGAGAGTTCCGGC-1_2 TTTGTTGCAAGTTCGT-1_2
# colData names(10): orig.ident nCount_RNA ... celltype ident
# reducedDimNames(4): PCA HARMONY UMAP TSNE
# mainExpName: RNA
# altExpNames(0):
# nhoods dimensions(2): 1 1
# nhoodCounts dimensions(2): 1 1
# nhoodDistances dimension(1): 0
# graph names(0):
# nhoodIndex names(1): 0
# nhoodExpression dimension(2): 1 1
# nhoodReducedDim names(0):
# nhoodGraph names(0):
# nhoodAdjacency dimension(2): 1 14. Построить K граф ближайших соседей.
# Значение d соответствует значению PCA, выбранному во время уменьшения размерности.
# Значение k соответствует значению, выбранному при использовании FindNeighbors.
reducedDims(scRNA_pre)
# List of length 4
# names(4): PCA HARMONY UMAP TSNE
scRNA_pre <- buildGraph(scRNA_pre, k = 15, d = 15,reduced.dim = "PCA")5. Определите репрезентативные окрестности на графе KNN.
# Определите значение точки данных (Prop)
# Для наборов данных размером менее 30 000 ячеек Prop устанавливается на 0,1.
# Для наборов данных, содержащих более 30 000 ячеек, свойство prop устанавливается равным 0,05.
scRNA_pre <- makeNhoods(scRNA_pre, prop = 0.1, k = 15, d = 15,
refined = TRUE, reduced_dims = "PCA")
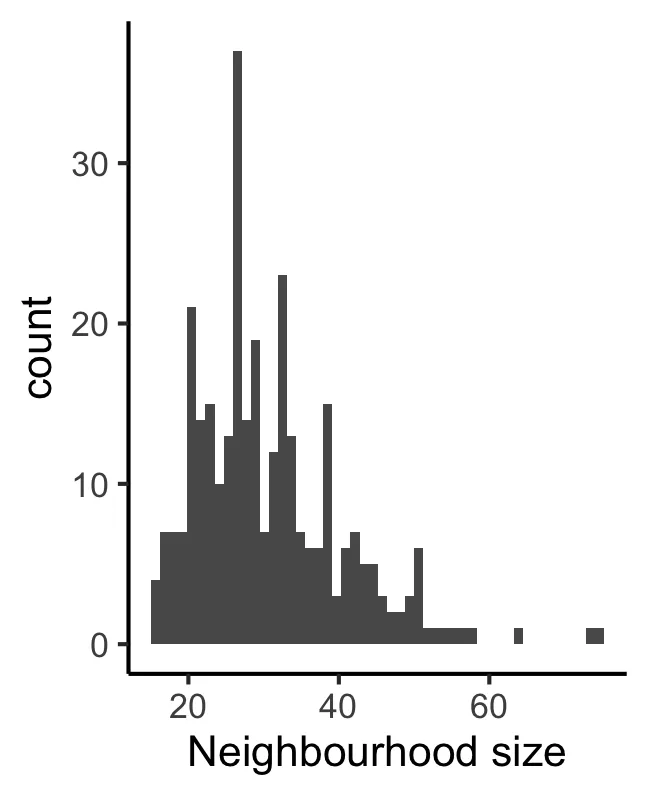
plotNhoodSizeHist(scRNA_pre)
Размер каждой определенной здесь окрестности составляет не менее 5 ячеек/выборку (личное понимание автора)
6. Подсчитайте ячейки в окрестностях
scRNA <- countCells(scRNA,
meta.data = as.data.frame(colData(scRNA)),
sample="orig.ident")
# colData(scRNA_pre) Эквивалент информации, представляющей метаданные
# образец представляет количество образцов
head(nhoodCounts(scRNA))
# На этом этапе можно добавить матрицу n*m к данным scRNA_pre.
# n представляет количество различных районов, m представляет собой экспериментальную выборку
# 6 x 6 sparse Matrix of class "dgCMatrix"
# sample1 sample2 sample3 sample4 sample5 sample6
# 1 . 10 . 7 20 9
# 2 . 17 . 6 6 4
# 3 22 2 50 . . 1
# 4 20 2 18 1 1 7
# 5 2 6 6 7 1 5
# 6 5 9 3 . . 57. Определение экспериментального плана
# Автор здесь пакет для информации о пакете
scRNA_design <- data.frame(colData(scRNA))[,c("orig.ident","group","batch")]
## Преобразовать информацию о пакете в целое число
scRNA_design$batch <- as.factor(scRNA_design$batch)
scRNA_design <- distinct(scRNA_design)
rownames(scRNA_design) <- scRNA_design$orig.ident
scRNA_design
# orig.ident group batch
# sample1 sample1 con 2
# sample2 sample2 con 1
# sample3 sample3 con 2
# sample4 sample4 treat 1
# sample5 sample5 treat 2
# sample6 sample6 treat 18. Рассчитайте связность района
# MiloR использует пространственную коррекцию FDR, введенную cydar. Этот шаг может исправить p-значения перекрытия между соседями.
scRNA <- calcNhoodDistance(scRNA, d=15, reduced.dim = "PCA")9.Результаты испытаний
# Добавление информации о партиях и группах в проект
da_results <- testNhoods(scRNA,design = ~ batch + group, design.df = scRNA_design)
head(da_results)
# logFC logCPM F PValue FDR Nhood SpatialFDR
# 1 3.1406854 12.86784 1.053518712 0.3049667 0.7636565 1 0.9681699
# 2 1.5241056 12.40488 0.463991996 0.4959349 0.9371039 2 0.9681699
# 3 -3.9628982 12.75593 1.284826869 0.2572975 0.7258632 3 0.9681699
# 4 -1.0256918 12.35456 0.131264086 0.7172078 0.9382545 4 0.9681699
# 5 -0.1763181 12.06026 0.008314397 0.9273667 0.9649626 5 0.9681699
# 6 -2.5291223 11.67320 1.547069145 0.2138841 0.6816075 6 0.9681699
da_results %>%
arrange(SpatialFDR) %>%
head()
ggplot(da_results, aes(PValue)) + geom_histogram(bins=50)
ggplot(da_results, aes(logFC, -log10(SpatialFDR))) +
geom_point() +
geom_hline(yintercept = 1) ## Mark significance threshold (10% FDR)Гистограмма P-значений: показывает распределение P-значений. Обычно в тестах дифференциальной численности (таких как MiloR) значение P измеряет статистическую значимость каждого района. Если значение P близко к 0, это означает, что окрестность между двумя условиями значительно различается. Как видно из рисунка, количество окрестностей со значениями P, близкими к 0, является самым большим, но значения P распределены относительно равномерно. Это показывает, что большинство районов не являются статистически значимыми, а некоторые имеют низкие значения P (т.е. существенные различия).
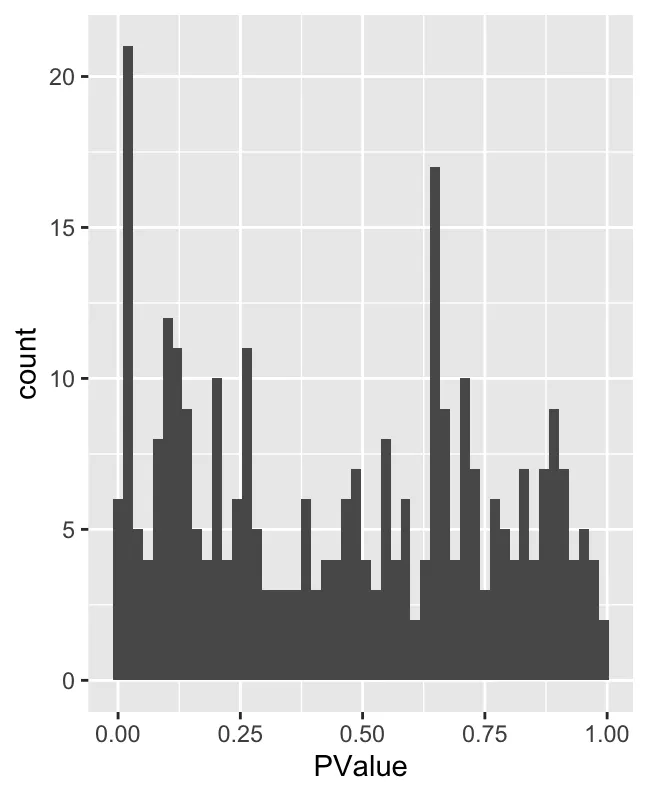
График рассеяния logFC и -log10(SpatialFDR): график рассеяния в стиле вулканической диаграммы с logFC на оси X и -log10(SpatialFDR) на оси Y. logFC означает логарифмическое изменение и используется для представления относительного изменения численности района в различных группах (таких как угощение и мошенничество). Положительные значения указывают на то, что район обогащается в группе лечения, а отрицательные значения указывают на то, что район обогащается в контрольной группе. SpatialFDR — это FDR с пространственной коррекцией. При построении графика -log10(SpatialFDR) чем выше значение, тем сильнее значимость (поскольку FDR меньше). Подавляющее большинство точек расположено ниже по оси Y, что указывает на то, что эти окрестности имеют высокие (т. е. незначительные) значения SpatialFDR. Другими словами, тест обнаружил очень мало дифференциальных соседей, что указывает на отсутствие сильного сигнала дифференциальной численности в данных.
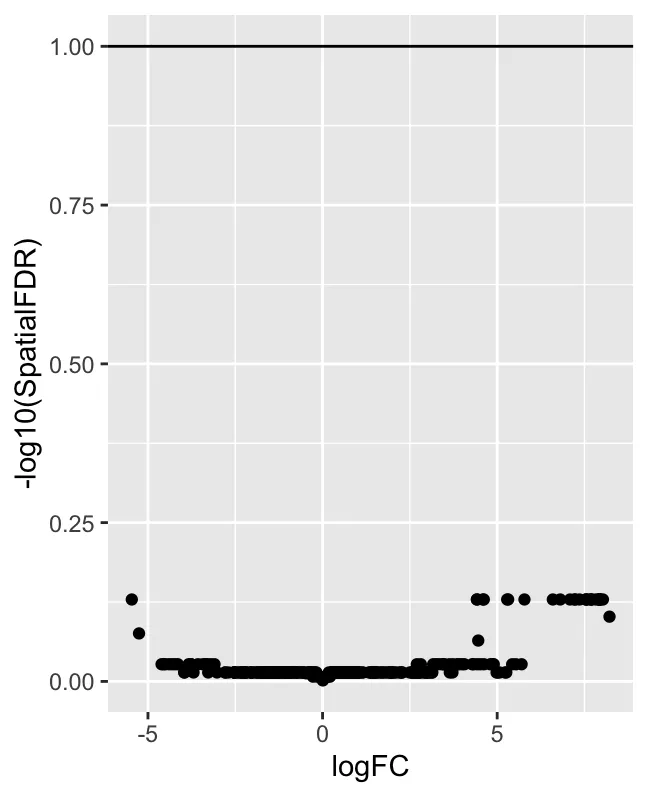
10.Визуализация
scRNA <- buildNhoodGraph(scRNA)
## Plot single-cell UMAP
umap_pl <- plotReducedDim(scRNA, dimred = "UMAP",
colour_by="group", text_by = "celltype",
text_size = 3, point_size=0.5) + guides(fill="none")
umap_pl
## Plot neighbourhood graph
nh_graph_pl <- plotNhoodGraphDA(scRNA, da_results,
layout="UMAP",alpha=0.9) #альфа по умолчанию 0,1
umap_pl + nh_graph_pl +
plot_layout(guides="collect")График UMAP (слева): показывает распределение различных типов клеток между двумя экспериментальными группами. На рисунке использованы два цвета: синий (против) и оранжевый (лечение), для обозначения контрольной группы и группы лечения соответственно. Различные типы клеток четко обозначены (например, фибробласты, B-клетки, моноциты и т. д.), и можно увидеть распределение клеток между обработанными (оранжевый) и контрольной (синий) группами.
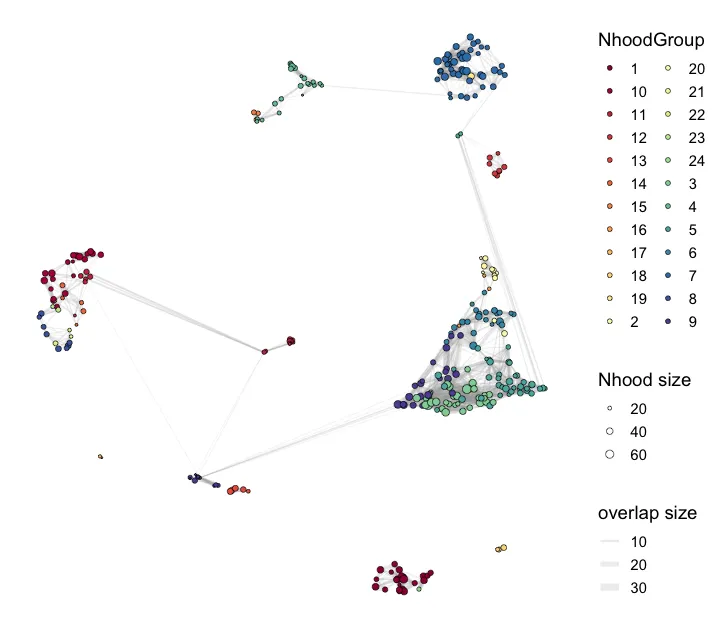
Граф сети окружения (справа). График справа представляет собой граф соседства, основанный на анализе MiloR. Каждый круг представляет собой окрестность (Nhood), а размер окрестности пропорционален количеству ячеек в ней. Цветовой градиент (от синего к красному) представляет logFC (логарифмическое изменение): положительный logFC (синий) означает, что клетки в этом районе обогащаются контрольной группой, отрицательный logFC (красный) указывает, что ячейки в окрестностях обогащается в группе лечения. Соединительные линии между районами представляют собой сходство или перекрытие между районами. Толщина линий представляет собой размер перекрытия между районами. Чем толще линии, тем более похожи ячейки между двумя районами.
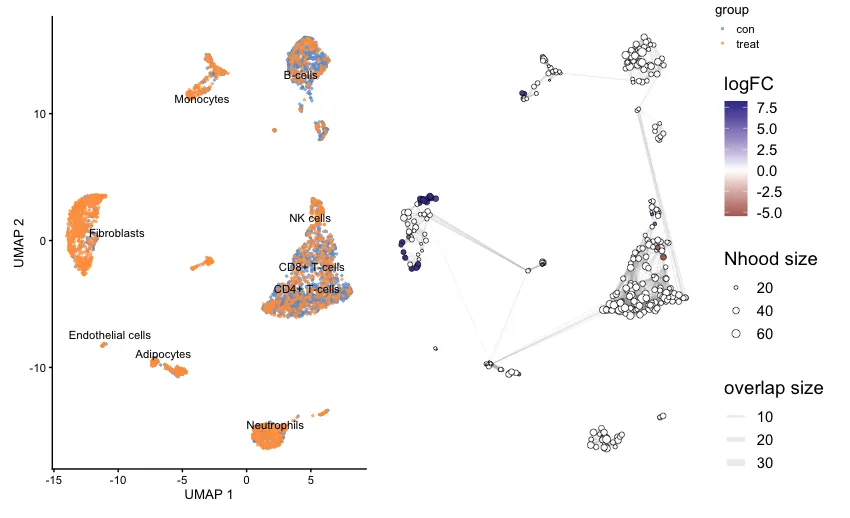
11.Аннотация данных
da_results <- annotateNhoods(scRNA,
da_results,
coldata_col = "celltype")
head(da_results)
# logFC logCPM F PValue FDR Nhood SpatialFDR celltype celltype_fraction
# 1 3.1406854 12.86784 1.053518712 0.3049667 0.7636565 1 0.9681699 Neutrophils 1.0000000
# 2 1.5241056 12.40488 0.463991996 0.4959349 0.9371039 2 0.9681699 NK cells 1.0000000
# 3 -3.9628982 12.75593 1.284826869 0.2572975 0.7258632 3 0.9681699 CD4+ T-cells 1.0000000
# 4 -1.0256918 12.35456 0.131264086 0.7172078 0.9382545 4 0.9681699 CD4+ T-cells 1.0000000
# 5 -0.1763181 12.06026 0.008314397 0.9273667 0.9649626 5 0.9681699 Monocytes 1.0000000
# 6 -2.5291223 11.67320 1.547069145 0.2138841 0.6816075 6 0.9681699 CD4+ T-cells 0.9545455
ggplot(da_results, aes(celltype_fraction)) + geom_histogram(bins=50)
str(da_results)
table(da_results$celltype)
range(da_results$SpatialFDR)
plotDAbeeswarm(da_results, group.by = "celltype",alpha = 0.9) # альфа по умолчанию равна 0,1Гистограмма celltype_fraction: показывает распределение пропорций определенного типа ячеек в окрестности (celltype_fraction). Ось Y представляет количество окрестностей, а ось X представляет долю типов ячеек в окрестности (в диапазоне от 0 до 1). Соотношение типов ячеек в большинстве районов близко к 1, что указывает на то, что в большинстве районов преобладает один тип ячеек. В большинстве районов преобладают ячейки определенного типа (celltype_fraction равен 1). Это означает, что клетки по соседству почти все одного типа, без явного смешения типов клеток. Этот результат обычно происходит в относительно чистых клеточных популяциях или клеточных субпопуляциях с четкими характеристиками разделения.
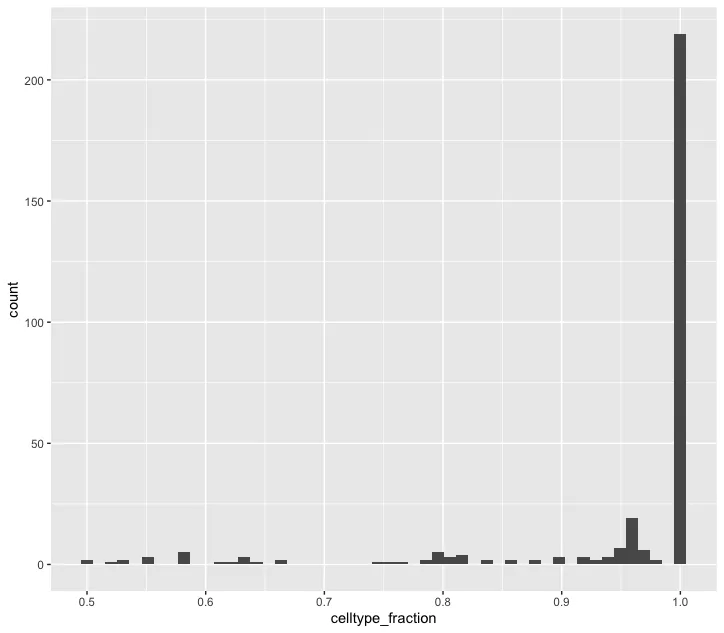
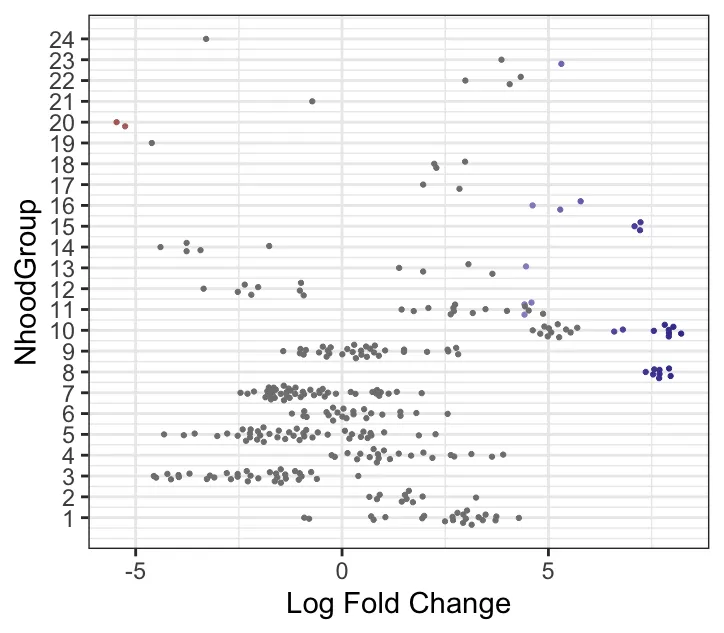
График зависимости logFC от типа ячейки: показывает распределение логарифмического кратного изменения (logFC) для разных типов клеток в разных окрестностях. Ось Y представляет тип клеток, а ось X представляет logFC (т.е. дифференциальные изменения численности в окрестностях между различными группами лечения). Каждая точка представляет собой окрестности, где разные цвета обозначают разные уровни значимости (отфильтрованные на основе FDR или p-значения). Очки для каждого типа ячейки отражают ее эффективность в разных районах. Типы клеток с более широким распределением значений logFC на рисунке, такие как фибробласты и CD8+ Т-клетки, демонстрируют значительные дифференциальные изменения численности между различными группами лечения. Некоторые типы клеток, такие как эндотелиальные клетки, могут иметь меньшие различия между группами. Большие значения logFC (например, больший прямой logFC) указывают на более высокую распространенность этих соседей в «лекарственной» группе. (Данные выборки автора не сильно отличаются)
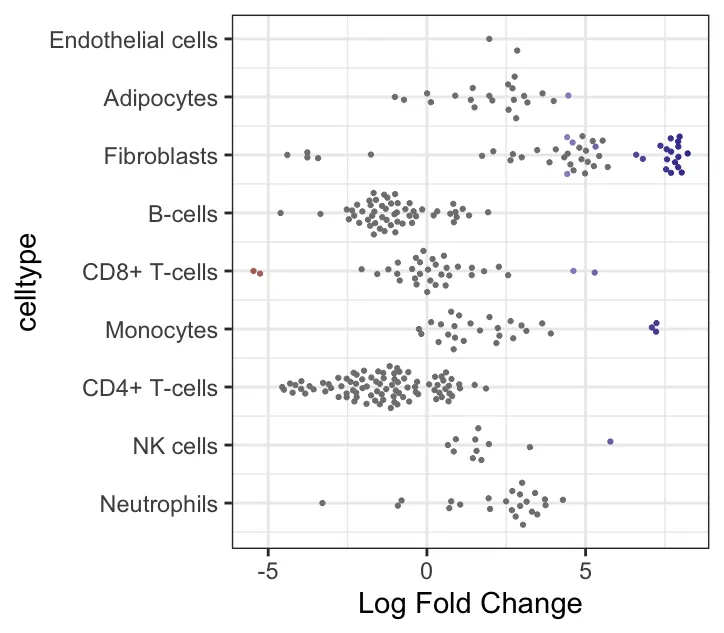
12. Найдите маркер группы DA.
## Add log normalized count to Milo object
scRNA <- logNormCounts(scRNA)
# альфа по умолчанию равна 0,1, поэтому SpatialFDR < 0.9 модифицирован
da_results$NhoodGroup <- as.numeric(da_results$SpatialFDR < 0.9 & da_results$logFC < 0)
da_nhood_markers <- findNhoodGroupMarkers(scRNA, da_results,
subset.row = rownames(scRNA),
aggregate.samples = TRUE, # Рекомендуется установить TRUE
sample_col = "orig.ident")
head(da_nhood_markers)
# GeneID logFC_0 adj.P.Val_0 logFC_1 adj.P.Val_1
# 1 AL627309.1 -0.0007221833 0.9413661 0.0007221833 0.9413661
# 2 AL627309.3 -0.0001256128 0.9413661 0.0001256128 0.9413661
# 3 AL645608.1 -0.0156444426 0.2788002 0.0156444426 0.2788002
# 4 AL669831.5 0.0100744381 0.9413661 -0.0100744381 0.9413661
# 5 FAM41C 0.0076732647 0.9413661 -0.0076732647 0.9413661
# 6 FAM87B -0.0056002509 0.3247399 0.0056002509 0.324739912.1. Автоматически группировать окрестности.
#Во многих случаях,Окрестности DA расположены в разных областях графа KNN.,все важно DA Группирование популяций вместе может быть не идеальным, поскольку они могут включать клетки самых разных типов клеток.
# Для этого сценария разработчик использовал функцию соседства, которая использует обнаружение сообществ для разделения районов на группы на основе (1) количества ячеек, общих для двух районов (2) DA; Направление изменения складок в сообществе; (3) разница в изменении складок;
## Run buildNhoodGraph to store nhood adjacency matrix
scRNA <- buildNhoodGraph(scRNA)
## Find groups
da_results <- groupNhoods(scRNA, da_results,
max.lfc.delta = 2,da.fdr = 0.9) #da.fdr по умолчанию равен 0, 1
head(da_results)
plotNhoodGroups(scRNA, da_results, layout="UMAP")
plotDAbeeswarm(da_results, "NhoodGroup",alpha = 0.9) #alpha по умолчанию — 0,1Этот шаг заключается в автоматическом разделении кластеров по результатам DA, а также позволяет найти дифференциальные гены. Значение max.lfc.delta можно изменить самостоятельно! В ходе формального анализа необходимы исследования.
12.2 Найдите характерные гены
## Exclude zero counts genes
keep.rows <- rowSums(logcounts(scRNA)) != 0
scRNA <- scRNA[keep.rows, ]
## Find HVGs
dec <- modelGeneVar(scRNA)
hvgs <- getTopHVGs(dec, n=2000)
head(hvgs)
# [1] "CFD" "DCN" "MGP" "CXCL8" "MT2A" "S100A9"
# Найдите дифференциальную экспрессию генов «один против всех» для каждой группы соседей.
nhood_markers <- findNhoodGroupMarkers(scRNA, da_results, subset.row = hvgs,
aggregate.samples = TRUE, sample_col = "orig.ident")
head(nhood_markers)[1:4,1:4]
# GeneID logFC_1 adj.P.Val_1 logFC_2
# 1 A2M -0.43376837 0.35815985 -0.43132559
# 2 AAK1 -0.55965379 0.03462636 0.74914069
# 3 ABCA1 0.20200482 0.46368550 -0.31103802
# 4 ABCA10 -0.08910405 0.50766539 -0.09984058
# Найдите дифференциальные гены группы2.
gr2_markers <- nhood_markers[c("logFC_2", "adj.P.Val_2")]
colnames(gr2_markers) <- c("logFC", "adj.P.Val")
head(gr2_markers[order(gr2_markers$adj.P.Val), ])
# Если понятно, что вас интересует вторая группа ячеек
nhood_markers <- findNhoodGroupMarkers(scRNA, da_results, subset.row = hvgs,
aggregate.samples = TRUE, sample_col = "orig.ident",
subset.groups = c("2")
)
head(nhood_markers)
# logFC_2 adj.P.Val_2 GeneID
# GNLY 4.529267 9.977073e-54 GNLY
# CLIC3 1.273255 4.886386e-40 CLIC3
# PRF1 2.186990 4.886386e-40 PRF1
# KLRF1 1.310507 2.133371e-35 KLRF1
# KLRD1 2.746359 9.425077e-32 KLRD1
# GZMB 2.731049 3.720297e-29 GZMB
# Если указана окрестность сравнения
nhood_markers <- findNhoodGroupMarkers(scRNA, da_results, subset.row = hvgs,
subset.nhoods = da_results$NhoodGroup %in% c('11','2'),
aggregate.samples = TRUE, sample_col = "orig.ident")
head(nhood_markers)
# GeneID logFC_2 adj.P.Val_2 logFC_11 adj.P.Val_11
# 1 A2M -1.1629210 0.004322766 1.1629210 0.004322766
# 2 AAK1 0.6881532 0.038952082 -0.6881532 0.038952082
# 3 ABCA1 -0.2253056 0.063886117 0.2253056 0.063886117
# 4 ABCA10 -0.1581780 0.064434029 0.1581780 0.064434029
# 5 ABCA6 -0.3517706 0.019652787 0.3517706 0.019652787
# 6 ABCA7 -0.1191482 0.307535525 0.1191482 0.30753552512.3. Визуализация дифференциальных генов
ggplot(nhood_markers, aes(logFC_2,-log10(adj.P.Val_2 ))) +
geom_point(alpha=0.5, size=0.5) +
geom_hline(yintercept = 2)
# Критерии фильтрации можно изменить самостоятельно.
markers <- rownames(nhood_markers)[nhood_markers$adj.P.Val_2 < 0.01 & nhood_markers$logFC_2 > 0]
plotNhoodExpressionGroups(scRNA, da_results, features=markers,
subset.nhoods = da_results$NhoodGroup %in% c('11','2'),
scale=TRUE,
grid.space = "fixed")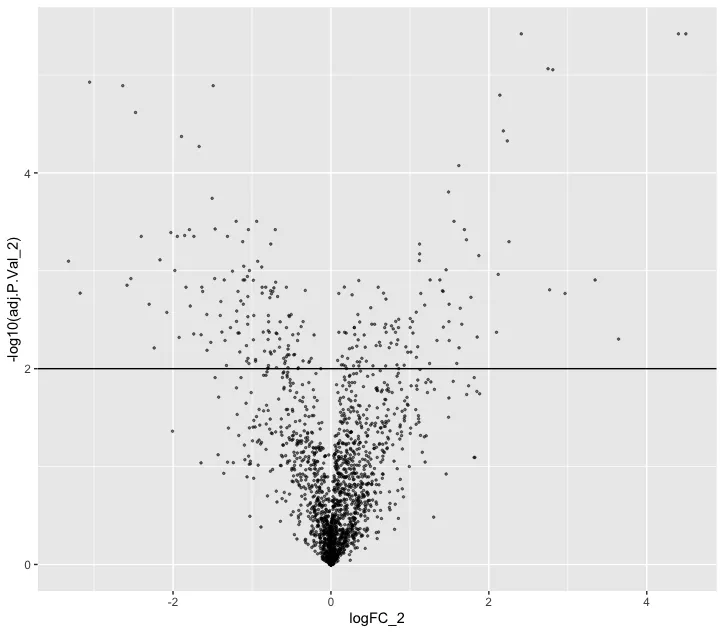
12.4 Сравнение ячеек в одном районе, но с разными вмешательствами
dge_9 <- testDiffExp(scRNA, da_results, design = ~ group,
meta.data = data.frame(colData(scRNA)),
subset.row = rownames(scRNA),
subset.nhoods=da_results$NhoodGroup=="9")
dge_9
# $`9`
# logFC AveExpr t P.Value adj.P.Val B Nhood.Group
# KLHL17 0.017612117 0.011781245 1.4215277 0.1558631 0.6982049 -9.789123 9
# LINC00115 0.026119993 0.041024269 1.1226770 0.2621795 0.6982049 -10.168746 9
# AL627309.1 0.002904512 0.001411100 1.0242142 0.3062900 0.6982049 -10.274403 9
# SAMD11 -0.006652596 0.003305143 -1.0015076 0.3171252 0.6982049 -10.297400 9
# NOC2L -0.030967073 0.180208912 -0.9373189 0.3491025 0.6982049 -10.359634 9
# AL669831.5 -0.005367254 0.007952223 -0.5865251 0.5578198 0.9296997 -10.627143 9
# FAM41C -0.002584961 0.014220774 -0.1830747 0.8548227 1.0000000 -10.782673 9
# AL627309.3 0.000000000 0.000000000 0.0000000 1.0000000 1.0000000 -10.799468 9
# FAM87B 0.000000000 0.000000000 0.0000000 1.0000000 1.0000000 -10.799468 9
# AL645608.1 0.000000000 0.000000000 0.0000000 1.0000000 1.0000000 -10.799468 9Напоминаю, что многие параметры модифицированы и аннотированы автором. При их использовании начинать анализ нужно со значений по умолчанию!
Ссылки:
- Differential abundance testing on single-cell data using k-nearest neighbor graphs. Nat Biotechnol. 2022 Feb;40(2):245-253.
- miloR:https://rawcdn.githack.com/MarioniLab/miloR/7c7f906b94a73e62e36e095ddb3e3567b414144e/vignettes/milo_gastrulation.html#5_Finding_markers_of_DA_populations
- одна ячейкамир:https://mp.weixin.qq.com/s/yYTxb_kOjeRSkzzuUoI4mA
Примечание:Если у вас есть какие-либо сомнения относительно содержания или вы обнаружили явную ошибку,,Пожалуйста, свяжитесь с серверной частью(Добро пожаловать для общения)。更多内容可закрывать Примечание Официальный аккаунт:Ковчег рождения
- END -



Неразрушающее увеличение изображений одним щелчком мыши, чтобы сделать их более четкими артефактами искусственного интеллекта, включая руководства по установке и использованию.
Копикодер: этот инструмент отлично работает с Cursor, Bolt и V0! Предоставьте более качественные подсказки для разработки интерфейса (создание навигационного веб-сайта с использованием искусственного интеллекта).
Новый бесплатный RooCline превосходит Cline v3.1? ! Быстрее, умнее и лучше вилка Cline! (Независимое программирование AI, порог 0)

Разработав более 10 проектов с помощью Cursor, я собрал 10 примеров и 60 подсказок.
Я потратил 72 часа на изучение курсорных агентов, и вот неоспоримые факты, которыми я должен поделиться!

Идеальная интеграция Cursor и DeepSeek API
DeepSeek V3 снижает затраты на обучение больших моделей
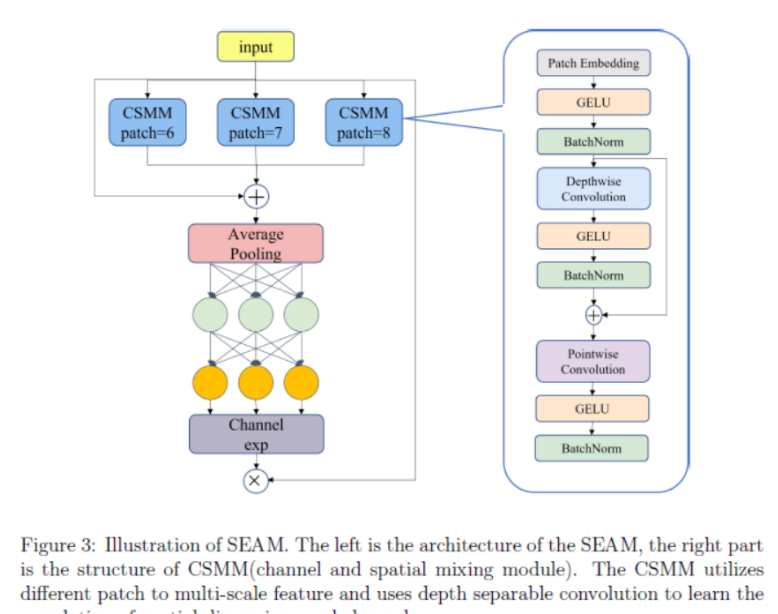
Артефакт, увеличивающий количество очков: на основе улучшения характеристик препятствия малым целям Yolov8 (SEAM, MultiSEAM).

DeepSeek V3 раскручивался уже три дня. Сегодня я попробовал самопровозглашенную модель «ChatGPT».

Open Devin — инженер-программист искусственного интеллекта с открытым исходным кодом, который меньше программирует и больше создает.
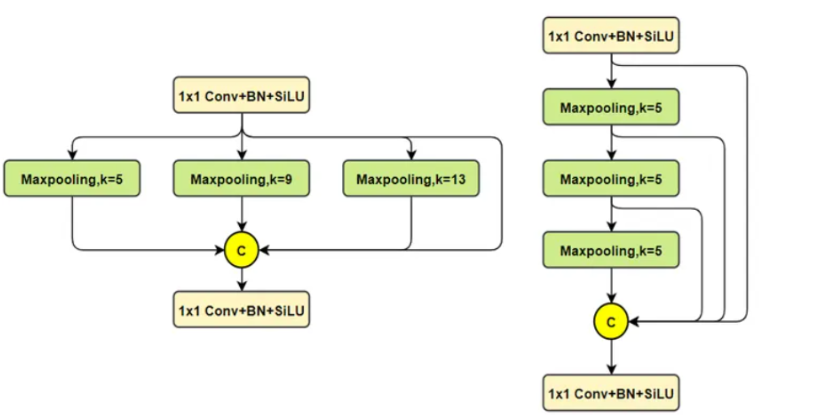
Эксклюзивное оригинальное улучшение YOLOv8: собственная разработка SPPF | SPPF сочетается с воспринимаемой большой сверткой ядра UniRepLK, а свертка с большим ядром + без расширения улучшает восприимчивое поле

Популярное и подробное объяснение DeepSeek-V3: от его появления до преимуществ и сравнения с GPT-4o.

9 основных словесных инструкций по доработке академических работ с помощью ChatGPT, эффективных и практичных, которые стоит собрать
Вызовите deepseek в vscode для реализации программирования с помощью искусственного интеллекта.
Познакомьтесь с принципами сверточных нейронных сетей (CNN) в одной статье (суперподробно)
50,3 тыс. звезд! Immich: автономное решение для резервного копирования фотографий и видео, которое экономит деньги и избавляет от беспокойства.
Cloud Native|Практика: установка Dashbaord для K8s, графика неплохая
Краткий обзор статьи — использование синтетических данных при обучении больших моделей и оптимизации производительности
MiniPerplx: новая поисковая система искусственного интеллекта с открытым исходным кодом, спонсируемая xAI и Vercel.
Конструкция сервиса Synology Drive сочетает проникновение в интрасеть и синхронизацию папок заметок Obsidian в облаке.
Центр конфигурации————Накос
Начинаем с нуля при разработке в облаке Copilot: начать разработку с минимальным использованием кода стало проще

[Серия Docker] Docker создает мультиплатформенные образы: практика архитектуры Arm64

Обновление новых возможностей coze | Я использовал coze для создания апплета помощника по исправлению домашних заданий по математике

Советы по развертыванию Nginx: практическое создание статических веб-сайтов на облачных серверах
Feiniu fnos использует Docker для развертывания личного блокнота Notepad

Сверточная нейронная сеть VGG реализует классификацию изображений Cifar10 — практический опыт Pytorch
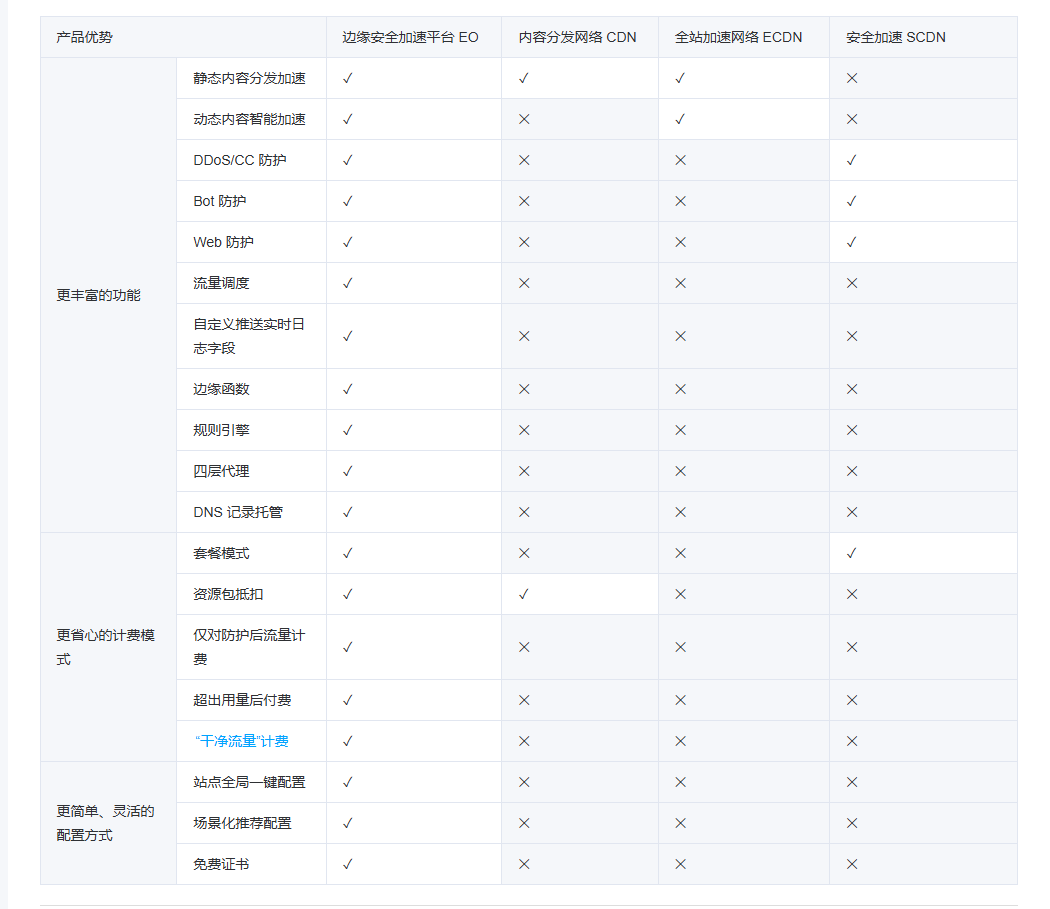
Начало работы с EdgeonePages — новым недорогим решением для хостинга веб-сайтов
[Зона легкого облачного игрового сервера] Управление игровыми архивами